当前位置:
X-MOL 学术
›
Dalton Trans.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Theoretical predictions of aromatic Be–O rings†
Dalton Transactions ( IF 4 ) Pub Date : 2018-08-13 00:00:00 , DOI: 10.1039/c8dt02020e Jason L. Dutton 1, 2, 3, 4, 5 , David J. D. Wilson 1, 2, 3, 4, 5
Dalton Transactions ( IF 4 ) Pub Date : 2018-08-13 00:00:00 , DOI: 10.1039/c8dt02020e Jason L. Dutton 1, 2, 3, 4, 5 , David J. D. Wilson 1, 2, 3, 4, 5
Affiliation
|
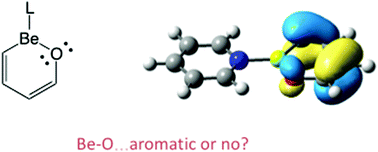
We have carried out a theoretical investigation of benzene substituted with BeO to assess stability and aromatic properties (with 1–3 BeO units in a six-membered ring); C4H4BeO, C2H2Be2O2, and Be3O3. All species retain a planar geometry. We considered both bare complexes and Be coordinated with L = CO, pyridine, PMe3, and NHC. Coordination of a ligand to Be atoms causes increased Be–O bond distances. Aromatic character has been investigated with NICS and NICS-scan calculations, EDA analysis, as well as considering the energetics of hydrogenation. The complexes with the greatest aromatic character are C4H4BeO (3), and C4H4Be(CO), which have negative NICS(1)zz values and minima in the NICS-scan. Coordination of two ligands to make the Be four-coordinate disrupts ring planarity and reduces aromatic character.
中文翻译:

芳香族Be–O环的理论预测†
我们已经对被BeO取代的苯进行了理论研究,以评估其稳定性和芳香性(六元环中具有1-3个BeO单元);C 4 H 4 BeO,C 2 H 2 Be 2 O 2和Be 3 O 3。所有种类均保留平面几何形状。我们既考虑了裸配合物,又与L = CO,吡啶,PMe 3和NHC配位。配体与Be原子的配位会导致Be-O键距离增加。芳香特性已通过NICS和NICS扫描计算,EDA分析以及考虑到氢化的能量学进行了研究。具有最大芳香特性的配合物是C4 H 4 BeO( 3)和C 4 H 4 Be(CO),它们在NICS扫描中具有负NICS(1) zz值和最小值。两个配体的配位以形成四配位,会破坏环的平面性并降低芳香性。
更新日期:2018-08-13
中文翻译:
芳香族Be–O环的理论预测†
我们已经对被BeO取代的苯进行了理论研究,以评估其稳定性和芳香性(六元环中具有1-3个BeO单元);C 4 H 4 BeO,C 2 H 2 Be 2 O 2和Be 3 O 3。所有种类均保留平面几何形状。我们既考虑了裸配合物,又与L = CO,吡啶,PMe 3和NHC配位。配体与Be原子的配位会导致Be-O键距离增加。芳香特性已通过NICS和NICS扫描计算,EDA分析以及考虑到氢化的能量学进行了研究。具有最大芳香特性的配合物是C4 H 4 BeO( 3)和C 4 H 4 Be(CO),它们在NICS扫描中具有负NICS(1) zz值和最小值。两个配体的配位以形成四配位,会破坏环的平面性并降低芳香性。
























 京公网安备 11010802027423号
京公网安备 11010802027423号