Organic Electronics ( IF 3.2 ) Pub Date : 2018-06-30 , DOI: 10.1016/j.orgel.2018.06.041 Yanan Zhu , Xiuru Xu , Xuepeng Zhang , Yaowu He , Xianzhe Zeng , Imran Murtaza , Hong Meng
|
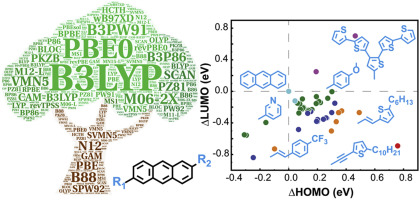
Anthracene derivatives constitute the active elements in high performance organic semiconductors and in order to find ubiquitous electronics applications, the investigation of new materials with high stability and mobilities is critical. Despite the versatility of substituent groups in anthracene and the extensive research works on anthracene derivatives, the exploration and discovery of high-performance anthracene derivatives can be impeded by the difficulty in molecular designing and synthesis techniques. Theoretical modeling is a powerful instrument and a variety of modeling methods with different accuracy levels have been reported. However, there is no systematic research work on theoretical prediction of the fundamental properties of anthracene derivatives so far. Here, density functional theory calculations are performed on anthracene derivatives to systematically address the prediction accuracy in energy levels as compared to our experimental data and it is shown that the PBE0/6-311G(d,p) is a more cost-effective method. Based on the influence of diverse substituent groups on energy levels, we present a strategy to design high-performance anthracene derivatives with specific energy levels, which are predicted to be excellent candidates for charge transfer materials. Therefore, the strategy may offer a guide for tailoring materials' functional properties via modifications of the molecular units before materials synthesis and open up new possibilities for the discovery of rising organic semiconductors.
中文翻译:
蒽基半导体的计算筛选和分子设计
蒽衍生物构成了高性能有机半导体中的活性元素,为了找到普遍存在的电子应用,研究具有高稳定性和迁移率的新材料至关重要。尽管蒽基中取代基的通用性和对蒽衍生物的广泛研究工作,但由于分子设计和合成技术的困难,高性能蒽衍生物的探索和发现受到了阻碍。理论建模是一种功能强大的工具,并且已经报道了各种具有不同准确度级别的建模方法。但是,迄今为止,对蒽衍生物的基本特性的理论预测尚无系统的研究。这里,与我们的实验数据相比,对蒽衍生物进行密度泛函理论计算以系统地解决能级的预测准确性,结果表明PBE0 / 6-311G(d,p)是一种更具成本效益的方法。基于不同取代基对能级的影响,我们提出了一种设计具有特定能级的高性能蒽衍生物的策略,预计该衍生物是电荷转移材料的极佳候选者。因此,该策略可为在材料合成之前通过分子单元的修饰来定制材料的功能特性提供指导,并为发现新兴的有机半导体开辟了新的可能性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号