当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Mechanisms and Active Sites for C–O Bond Rupture within 2-Methyltetrahydrofuran over Ni, Ni12P5, and Ni2P Catalysts
ACS Catalysis ( IF 12.9 ) Pub Date : 2018-06-25 00:00:00 , DOI: 10.1021/acscatal.7b04403 Megan E. Witzke 1 , Abdulrahman Almithn 2 , Christian L. Conrad 1 , David D. Hibbitts 2 , David W. Flaherty 1
ACS Catalysis ( IF 12.9 ) Pub Date : 2018-06-25 00:00:00 , DOI: 10.1021/acscatal.7b04403 Megan E. Witzke 1 , Abdulrahman Almithn 2 , Christian L. Conrad 1 , David D. Hibbitts 2 , David W. Flaherty 1
Affiliation
|
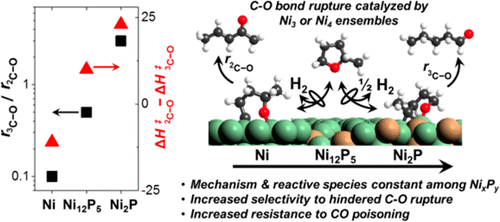
Nickel phosphide catalysts (Ni12P5 and Ni2P) preferentially cleave sterically hindered 3C–O bonds over unhindered 2C–O bonds, and Ni2P is up to 50 times more selective toward 3C–O bond cleavage than Ni. Here, we combine kinetic measurements, in situ infrared spectroscopy, and density functional theory (DFT) calculations to describe the mechanism for C–O bond rupture over Ni, Ni12P5, and Ni2P catalysts. Steady-state rate measurements and DFT calculations of C–O bond rupture within 2-methyltetrahydrofuran (MTHF) show that quasi-equilibrated MTHF adsorption and dehydrogenation steps precede kinetically relevant C–O bond rupture at these conditions (1–50 kPa MTHF; 0.1–6 MPa H2; 543 K). Rates for 3C–O and 2C–O bond rupture are inhibited by H2, and the ratio of these rates increases with [H2]1/2, suggesting that the composition of the reactive intermediates for 3C–O and 2C–O rupture differs by one H atom. Site-blocking CO*, NH3*, and H* inhibit rates without altering the ratio of 3C–O to 2C–O bond rupture, indicating that these C–O bond rupture precursors and transition states bind to identical active sites. DFT-based predictions suggest that these sites are exposed ensembles of 3 Ni atoms on Ni(111) and Ni2P(001) and 4 Ni atoms on Ni12P5(001) and that the incorporation of P disrupts extended Ni ensembles and alters the reactivity of the Ni. Increasing the phosphorus to nickel ratio (P:Ni) decreases measured and DFT-predicted activation enthalpies (ΔH‡, 473–583 K) for 3C–O bond rupture relative to that of 2C–O bond rupture. Selectivity differences between specific C–O bonds within MTHF reflect differences in the H content of reactive intermediates, activation enthalpy barriers, and P:Ni of Ni, Ni12P5, and Ni2P nanoparticles.
中文翻译:

Ni,Ni 12 P 5和Ni 2 P催化剂上2-甲基四氢呋喃中C–O键断裂的机理和活性中心
磷化镍催化剂(Ni 12 P 5和Ni 2 P)优先裂解空间受阻的3 C-O键,而不是不受阻碍的2 C-O键,并且Ni 2 P对3 C-O键断裂的选择性是Ni的50倍以上。在这里,我们结合动力学测量,原位红外光谱和密度泛函理论(DFT)计算来描述Ni,Ni 12 P 5和Ni 2上C–O键断裂的机理。磷催化剂。稳态速率测量和2-甲基四氢呋喃(MTHF)中C-O键断裂的DFT计算表明,在这些条件下,准平衡的MTHF吸附和脱氢步骤先于动力学相关的C-O键断裂(1-50 kPa MTHF; 0.1 –6 MPa H 2; 543 K)。对于价格3 C-O和2 C-O键断裂是由H抑制2,以及它们的速率随[H的比率增加2 ] 1/2,表明活性中间体的组合物3 C-O和2 C–O断裂相差一个H原子。阻止位点的CO *,NH 3 *和H *抑制速率而不改变3的比例C–O至2个C–O键断裂,表明这些C–O键断裂的前体和过渡态与相同的活性位结合。基于DFT的预测表明这些位点是Ni(111)和Ni 2 P(001)上的3个Ni原子和Ni 12 P 5(001)上的4个Ni原子的暴露集合,并且P的掺入破坏了扩展的Ni集合。改变镍的反应性 增加的磷镍比(P:Ni)降低了,对于3 C-O键断裂,DFT预测的活化焓(ΔH ‡,473-583 K)相对于2C–O键断裂。MTHF中特定C–O键之间的选择性差异反映了反应性中间体H含量,活化焓屏障以及Ni,Ni 12 P 5和Ni 2 P纳米粒子的P:Ni的差异。
更新日期:2018-06-25
中文翻译:
Ni,Ni 12 P 5和Ni 2 P催化剂上2-甲基四氢呋喃中C–O键断裂的机理和活性中心
磷化镍催化剂(Ni 12 P 5和Ni 2 P)优先裂解空间受阻的3 C-O键,而不是不受阻碍的2 C-O键,并且Ni 2 P对3 C-O键断裂的选择性是Ni的50倍以上。在这里,我们结合动力学测量,原位红外光谱和密度泛函理论(DFT)计算来描述Ni,Ni 12 P 5和Ni 2上C–O键断裂的机理。磷催化剂。稳态速率测量和2-甲基四氢呋喃(MTHF)中C-O键断裂的DFT计算表明,在这些条件下,准平衡的MTHF吸附和脱氢步骤先于动力学相关的C-O键断裂(1-50 kPa MTHF; 0.1 –6 MPa H 2; 543 K)。对于价格3 C-O和2 C-O键断裂是由H抑制2,以及它们的速率随[H的比率增加2 ] 1/2,表明活性中间体的组合物3 C-O和2 C–O断裂相差一个H原子。阻止位点的CO *,NH 3 *和H *抑制速率而不改变3的比例C–O至2个C–O键断裂,表明这些C–O键断裂的前体和过渡态与相同的活性位结合。基于DFT的预测表明这些位点是Ni(111)和Ni 2 P(001)上的3个Ni原子和Ni 12 P 5(001)上的4个Ni原子的暴露集合,并且P的掺入破坏了扩展的Ni集合。改变镍的反应性 增加的磷镍比(P:Ni)降低了,对于3 C-O键断裂,DFT预测的活化焓(ΔH ‡,473-583 K)相对于2C–O键断裂。MTHF中特定C–O键之间的选择性差异反映了反应性中间体H含量,活化焓屏障以及Ni,Ni 12 P 5和Ni 2 P纳米粒子的P:Ni的差异。
























 京公网安备 11010802027423号
京公网安备 11010802027423号