当前位置:
X-MOL 学术
›
Mol. Pharmaceutics
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Aceclofenac–Galactose Conjugate: Design, Synthesis, Characterization, and Pharmacological and Toxicological Evaluations
Molecular Pharmaceutics ( IF 4.9 ) Pub Date : 2018-06-18 00:00:00 , DOI: 10.1021/acs.molpharmaceut.8b00195 Salvatore Magliocca 1 , Carmen De Caro 1, 2 , Loretta Lazzarato , Roberto Russo 1 , Barbara Rolando , Konstantin Chegaev , Elisabetta Marini , Maria Nieddu 3 , Lucia Burrai 3 , Gianpiero Boatto 3 , Claudia Cristiano 1 , Domenica Marabello 4 , Elena Gazzano 5 , Chiara Riganti 5 , Federica Sodano , Maria Grazia Rimoli 1
Molecular Pharmaceutics ( IF 4.9 ) Pub Date : 2018-06-18 00:00:00 , DOI: 10.1021/acs.molpharmaceut.8b00195 Salvatore Magliocca 1 , Carmen De Caro 1, 2 , Loretta Lazzarato , Roberto Russo 1 , Barbara Rolando , Konstantin Chegaev , Elisabetta Marini , Maria Nieddu 3 , Lucia Burrai 3 , Gianpiero Boatto 3 , Claudia Cristiano 1 , Domenica Marabello 4 , Elena Gazzano 5 , Chiara Riganti 5 , Federica Sodano , Maria Grazia Rimoli 1
Affiliation
|
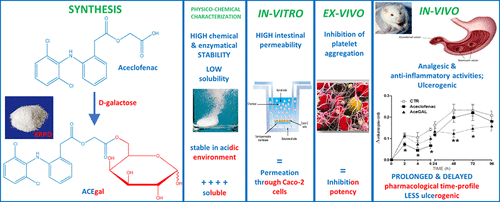
Aceclofenac is a popular analgesic, antipyretic, and nonsteroidal anti-inflammatory drug (NSAID) used for prolonged treatment (at least three months) in musculoskeletal disorders. It is characterized by several limitations such as poor water solubility and low oral bioavailability. The main side-effect of aceclofenac, as well as all NSAIDs, is the gastrotoxicity; among other adverse effects, there is the risk of bleeding since aceclofenac reversibly inhibits platelet aggregation. With the aim to reduce these drawbacks, we have designed, synthesized, and characterized, both in vitro and in vivo, an orally administrable pro-drug of aceclofenac (ACEgal). ACEgal was obtained by conjugating carboxyl group with the 6-OH group of d-galactose; its structure was confirmed by X-ray powder diffractometry. The pro-drug was shown to be stable at 37 °C in simulated gastric fluid (SGF-without pepsin, pH = 1.2) and moderately stable in phosphate buffered saline (PBS, pH = 7.4). However, it hydrolyzed in human serum with a half-life (t1/2) of 36 min, producing aceclofenac. Furthermore, if compared to its parent drug, ACEgal was four-times more soluble in SGF. To predict human intestinal absorption, cell permeability in a Caco-2 model of aceclofenac and ACEgal was determined. Anti-inflammatory, analgesic, and ulcerogenic activities have been investigated in vivo. In addition, oxidative stress parameters (thiobarbituric acid reactive substances, TBARS, and glutathione, GSH) and platelet antiaggregatory activity both of parent drug and pro-drug were evaluated. Results clearly showed that the conjugation of aceclofenac to a galactose molecule improves physicochemical, toxicological (at gastric and blood level), and pharmacological profile of aceclofenac itself without changing intestinal permeability and antiplatelet activity (in spite the new sugar moiety).
中文翻译:

醋氯芬酸-半乳糖结合物:设计,合成,表征以及药理和毒理学评估
醋氯芬酸是一种流行的止痛,解热和非甾体抗炎药(NSAID),可用于肌肉骨骼疾病的延长治疗(至少三个月)。它的特点是水溶性差和口服生物利用度低等几个限制。醋氯芬酸以及所有非甾体抗炎药的主要副作用是胃肠毒性。除其他不利影响外,还有出血的风险,因为醋氯芬酸可逆地抑制血小板凝集。其目的,以减少这些缺点,我们已设计,合成,和其特征在于,无论是在体外和体内,醋氯芬酸的可口服给药的前体药物(ACEgal)。通过使羧基与d的6-OH基团共轭获得ACEgal-半乳糖; 其结构通过X射线粉末衍射法确认。该前药在模拟胃液(不含胃蛋白酶的SGF,pH = 1.2)中在37°C下稳定,在磷酸盐缓冲液(PBS,pH = 7.4)中中等稳定。但是,它在人血清中水解的半衰期(t 1/2)为36分钟,产生醋氯芬酸。此外,如果与母药相比,ACEgal在SGF中的溶解度要高四倍。为了预测人体的肠道吸收,确定了醋氯芬酸和ACEgal的Caco-2模型中的细胞通透性。在体内已经研究了抗炎,止痛和促溃疡活性。此外,还评估了母体药物和前体药物的氧化应激参数(硫代巴比妥酸反应性物质,TBARS和谷胱甘肽,GSH)和血小板的抗聚集活性。结果清楚地表明,醋氯芬酸与半乳糖分子的缀合可改善醋氯芬酸本身的理化,毒理学(在胃和血液水平)和药理学特性,而不会改变肠的通透性和抗血小板活性(尽管有新的糖部分)。
更新日期:2018-06-18
中文翻译:
醋氯芬酸-半乳糖结合物:设计,合成,表征以及药理和毒理学评估
醋氯芬酸是一种流行的止痛,解热和非甾体抗炎药(NSAID),可用于肌肉骨骼疾病的延长治疗(至少三个月)。它的特点是水溶性差和口服生物利用度低等几个限制。醋氯芬酸以及所有非甾体抗炎药的主要副作用是胃肠毒性。除其他不利影响外,还有出血的风险,因为醋氯芬酸可逆地抑制血小板凝集。其目的,以减少这些缺点,我们已设计,合成,和其特征在于,无论是在体外和体内,醋氯芬酸的可口服给药的前体药物(ACEgal)。通过使羧基与d的6-OH基团共轭获得ACEgal-半乳糖; 其结构通过X射线粉末衍射法确认。该前药在模拟胃液(不含胃蛋白酶的SGF,pH = 1.2)中在37°C下稳定,在磷酸盐缓冲液(PBS,pH = 7.4)中中等稳定。但是,它在人血清中水解的半衰期(t 1/2)为36分钟,产生醋氯芬酸。此外,如果与母药相比,ACEgal在SGF中的溶解度要高四倍。为了预测人体的肠道吸收,确定了醋氯芬酸和ACEgal的Caco-2模型中的细胞通透性。在体内已经研究了抗炎,止痛和促溃疡活性。此外,还评估了母体药物和前体药物的氧化应激参数(硫代巴比妥酸反应性物质,TBARS和谷胱甘肽,GSH)和血小板的抗聚集活性。结果清楚地表明,醋氯芬酸与半乳糖分子的缀合可改善醋氯芬酸本身的理化,毒理学(在胃和血液水平)和药理学特性,而不会改变肠的通透性和抗血小板活性(尽管有新的糖部分)。
























 京公网安备 11010802027423号
京公网安备 11010802027423号