当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Ab Initio Wave Function-Based Determination of Element Specific Shifts for the Efficient Calculation of X-ray Absorption Spectra of Main Group Elements and First Row Transition Metals
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2018-06-12 00:00:00 , DOI: 10.1021/acs.jctc.8b00249 Agisilaos Chantzis 1, 2 , Joanna K. Kowalska 1 , Dimitrios Maganas 1, 2 , Serena DeBeer 1 , Frank Neese 1, 2
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2018-06-12 00:00:00 , DOI: 10.1021/acs.jctc.8b00249 Agisilaos Chantzis 1, 2 , Joanna K. Kowalska 1 , Dimitrios Maganas 1, 2 , Serena DeBeer 1 , Frank Neese 1, 2
Affiliation
|
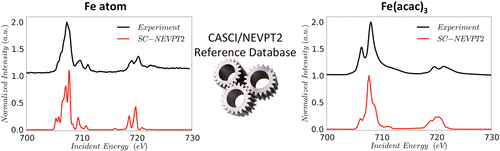
In this study, a detailed calibration of the performance of modern ab initio wave function methods in the domain of X-ray absorption spectroscopy (XAS) is presented. It has been known for some time that for a given level of approximation, for example, using time-dependent density functional theory (TD-DFT) in conjunction with a given basis set, there are systematic deviations of the calculated transition energies from their experimental values that depend on the functional, the basis set, and the chosen treatment of scalar relativistic effects. This necessitates a linear correlation for a given element/functional/basis set combination to be established before chemical applications can be performed. This is a laborious undertaking since it involves sourcing trustworthy experimental data, lengthy geometry optimizations, and detailed comparisons between theory and experiment. In this work, reference values for the element-specific shifts of all the first-row transition metal atoms and the main group elements C, N, O, F, Si, P, S, and Cl have been computed by using a protocol that is based on the complete active space configuration interaction in conjunction with second-order N-electron valence state perturbation theory (CASCI/NEVPT2). It is shown that by extrapolating the results to the basis set limit of the method and taking into account scalar relativistic effects via the second-order Douglas–Kroll–Hess (DKH2) corrections, the predicted element shifts are on average less than 0.75 eV across all the absorption edges and a very good agreement between theory and experiment in all the studied XAS cases is observed. The transferability of these errors to molecular systems is thoroughly investigated. The constructed CASCI/NEVPT2 database of element shifts is used to evaluate the performance and to automatically calibrate prior to comparison with the experiment two commonly used methods in X-ray spectroscopy, namely, DFT/Restricted open shell configuration interaction singles (DFT/ROCIS) and TD-DFT methods.
中文翻译:

基于从头算波函数的元素比位移的确定,可高效计算主族元素和第一行过渡金属的X射线吸收光谱
在这项研究中,对现代从头开始的性能进行了详细的校准介绍了X射线吸收光谱(XAS)领域中的波函数方法。一段时间以来,人们已经知道,对于给定的近似水平,例如,使用时变密度泛函理论(TD-DFT)并结合给定的基集,计算得出的跃迁能与实验值存在系统性偏差。值取决于函数,基集和标量相对论效应的选择处理方式。对于给定的元素/功能/基集组合,必须先进行线性相关,然后才能进行化学应用。这是一项艰巨的工作,因为它涉及到获得可信赖的实验数据,冗长的几何优化以及理论与实验之间的详细比较。在这项工作中,ñ-电子价态微扰理论(CASCI / NEVPT2)。结果表明,通过将结果外推到方法的基本极限,并考虑通过二阶道格拉斯-克罗尔-赫斯(DKH2)校正的标量相对论效应,在整个过程中,预测的元素位移平均小于0.75 eV。在所有研究的XAS情况下,都观察到了所有吸收边,并且理论与实验之间有很好的一致性。这些错误到分子系统的转移能力得到了彻底的研究。构建的CASCI / NEVPT2元素位移数据库用于评估性能并在与实验进行比较之前进行自动校准,这是X射线光谱中的两种常用方法,即DFT /受限开放壳结构相互作用单体(DFT / ROCIS)和TD-DFT方法。
更新日期:2018-06-12
中文翻译:
基于从头算波函数的元素比位移的确定,可高效计算主族元素和第一行过渡金属的X射线吸收光谱
在这项研究中,对现代从头开始的性能进行了详细的校准介绍了X射线吸收光谱(XAS)领域中的波函数方法。一段时间以来,人们已经知道,对于给定的近似水平,例如,使用时变密度泛函理论(TD-DFT)并结合给定的基集,计算得出的跃迁能与实验值存在系统性偏差。值取决于函数,基集和标量相对论效应的选择处理方式。对于给定的元素/功能/基集组合,必须先进行线性相关,然后才能进行化学应用。这是一项艰巨的工作,因为它涉及到获得可信赖的实验数据,冗长的几何优化以及理论与实验之间的详细比较。在这项工作中,ñ-电子价态微扰理论(CASCI / NEVPT2)。结果表明,通过将结果外推到方法的基本极限,并考虑通过二阶道格拉斯-克罗尔-赫斯(DKH2)校正的标量相对论效应,在整个过程中,预测的元素位移平均小于0.75 eV。在所有研究的XAS情况下,都观察到了所有吸收边,并且理论与实验之间有很好的一致性。这些错误到分子系统的转移能力得到了彻底的研究。构建的CASCI / NEVPT2元素位移数据库用于评估性能并在与实验进行比较之前进行自动校准,这是X射线光谱中的两种常用方法,即DFT /受限开放壳结构相互作用单体(DFT / ROCIS)和TD-DFT方法。

























 京公网安备 11010802027423号
京公网安备 11010802027423号