当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Toward Accurate QM/MM Reaction Barriers with Large QM Regions Using Domain Based Pair Natural Orbital Coupled Cluster Theory
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2018-06-08 00:00:00 , DOI: 10.1021/acs.jctc.8b00348 Giovanni Bistoni 1 , Iakov Polyak 1 , Manuel Sparta 1 , Walter Thiel 1 , Frank Neese 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2018-06-08 00:00:00 , DOI: 10.1021/acs.jctc.8b00348 Giovanni Bistoni 1 , Iakov Polyak 1 , Manuel Sparta 1 , Walter Thiel 1 , Frank Neese 1
Affiliation
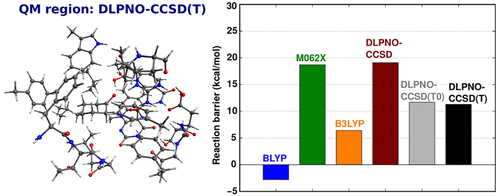
|
The hydroxylation reaction catalyzed by p-hydroxybenzoate hydroxylase and the Baeyer–Villiger reaction catalyzed by cyclohexanone monooxygenase are investigated by means of quantum mechanical/molecular mechanical (QM/MM) calculations at different levels of QM theory. The geometries of the stationary points along the reaction profile are obtained from QM/MM geometry optimizations, in which the QM region is treated by density functional theory (DFT). Relative energies are determined from single-point QM/MM calculations using the domain-based local pair natural orbital coupled cluster DLPNO-CCSD(T) method as QM component. The results are compared with single-point DFT/MM energies obtained using popular density functionals and with available experimental and computational data. It is found that the choice of the QM method strongly affects the computed energy profiles for these reactions. Different density functionals provide qualitatively different energy barriers (variations of the order of 10 kcal/mol in both reactions), thus limiting the confidence in DFT/MM computational predictions of energy profiles. On the other hand, the use of the DLPNO-CCSD(T) method in conjunction with large QM regions and basis sets makes it possible to achieve high accuracy. A critical discussion of all the technical aspects of the calculations is given with the aim of aiding computational chemists in the application of the DLPNO-CCSD(T) methodology in QM/MM calculations.
中文翻译:

利用基于域的成对自然轨道耦合簇理论向大QM区精确的QM / MM反应壁垒
p催化的羟基化反应通过量子力学/分子力学(QM / MM)计算方法研究了不同水平的QM理论中的-羟基苯甲酸酯羟化酶和环己酮单加氧酶催化的Baeyer-Villiger反应。沿着反应曲线的固定点的几何结构是从QM / MM几何结构优化中获得的,其中QM区域通过密度泛函理论(DFT)进行处理。使用基于域的本地对自然轨道耦合簇DLPNO-CCSD(T)方法作为QM分量,从单点QM / MM计算确定相对能量。将结果与使用流行的密度泛函获得的单点DFT / MM能量以及可用的实验和计算数据进行比较。发现,QM方法的选择强烈影响这些反应的计算能量分布。不同的密度泛函在质上提供了不同的能垒(两个反应的变化幅度均为10 kcal / mol),从而限制了对DFT / MM计算能量分布预测的信心。另一方面,将DLPNO-CCSD(T)方法与较大的QM区域和基集结合使用可以实现高精度。为了帮助计算化学家将DLPNO-CCSD(T)方法应用于QM / MM计算,对计算的所有技术方面进行了批判性讨论。将DLPNO-CCSD(T)方法与较大的QM区域和基集结合使用可以实现高精度。为了帮助计算化学家将DLPNO-CCSD(T)方法应用于QM / MM计算,对计算的所有技术方面进行了批判性讨论。将DLPNO-CCSD(T)方法与较大的QM区域和基集结合使用可以实现高精度。为了帮助计算化学家将DLPNO-CCSD(T)方法应用于QM / MM计算,对计算的所有技术方面进行了批判性讨论。
更新日期:2018-06-08
中文翻译:
利用基于域的成对自然轨道耦合簇理论向大QM区精确的QM / MM反应壁垒
p催化的羟基化反应通过量子力学/分子力学(QM / MM)计算方法研究了不同水平的QM理论中的-羟基苯甲酸酯羟化酶和环己酮单加氧酶催化的Baeyer-Villiger反应。沿着反应曲线的固定点的几何结构是从QM / MM几何结构优化中获得的,其中QM区域通过密度泛函理论(DFT)进行处理。使用基于域的本地对自然轨道耦合簇DLPNO-CCSD(T)方法作为QM分量,从单点QM / MM计算确定相对能量。将结果与使用流行的密度泛函获得的单点DFT / MM能量以及可用的实验和计算数据进行比较。发现,QM方法的选择强烈影响这些反应的计算能量分布。不同的密度泛函在质上提供了不同的能垒(两个反应的变化幅度均为10 kcal / mol),从而限制了对DFT / MM计算能量分布预测的信心。另一方面,将DLPNO-CCSD(T)方法与较大的QM区域和基集结合使用可以实现高精度。为了帮助计算化学家将DLPNO-CCSD(T)方法应用于QM / MM计算,对计算的所有技术方面进行了批判性讨论。将DLPNO-CCSD(T)方法与较大的QM区域和基集结合使用可以实现高精度。为了帮助计算化学家将DLPNO-CCSD(T)方法应用于QM / MM计算,对计算的所有技术方面进行了批判性讨论。将DLPNO-CCSD(T)方法与较大的QM区域和基集结合使用可以实现高精度。为了帮助计算化学家将DLPNO-CCSD(T)方法应用于QM / MM计算,对计算的所有技术方面进行了批判性讨论。

























 京公网安备 11010802027423号
京公网安备 11010802027423号