当前位置:
X-MOL 学术
›
Org. Chem. Front.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
The effects of different heterocycles and solvents on the ESIPT mechanisms of three novel photoactive mono-formylated benzoxazole derivatives†
Organic Chemistry Frontiers ( IF 5.4 ) Pub Date : 2018-06-09 00:00:00 , DOI: 10.1039/c8qo00349a Jiaojiao Hao 1, 2, 3, 4, 5 , Yang Yang Yang Yang 1, 2, 3, 4, 5
Organic Chemistry Frontiers ( IF 5.4 ) Pub Date : 2018-06-09 00:00:00 , DOI: 10.1039/c8qo00349a Jiaojiao Hao 1, 2, 3, 4, 5 , Yang Yang Yang Yang 1, 2, 3, 4, 5
Affiliation
|
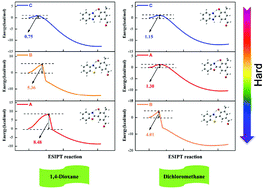
The fluorescence behaviors and properties of three novel photoactive mono-formylated benzoxazole derivatives A–C are found to be affected by different heterocycles and solvents, as reported in a recent experiment (Rodembusch, et al., New J. Chem., 2016, 40, 2785). Unfortunately, the detailed excited-state intramolecular proton transfer (ESIPT) mechanisms of these compounds are lacking. In this study, we used density functional theory (DFT) and time-dependent DFT (TDDFT) methods to study the dynamic ESIPT processes of the three compounds A–C in two different surroundings (polar 1,4-dioxane and nonpolar dichloromethane solvents). The calculated absorption and fluorescence spectra were observed to mutually agree with the experimental data, which indicated that the TDDFT method we adopted was reliable. In addition, based on the analysis of bond lengths, bond angles and IR vibrational spectra in both solvents, it was confirmed that the intramolecular hydrogen bonds (HBs) of these compounds were strengthened in the S1 state, which could promote the ESIPT reactions. Moreover, the frontier molecular orbitals (MOs) and the maps of the electron density difference between the S0 and S1 states displayed intramolecular charge transfer, which provided the probability of ESIPT reactions for the three compounds. Furthermore, based on the constructed potential energy curves, we revealed detailed dynamical ESIPT mechanisms of the compounds A–C. As a consequence, we found that the ESIPT processes were more likely to take place from A (8.48 kcal mol−1) → B (5.36 kcal mol−1) → C (0.75 kcal mol−1) compounds in the polar 1,4-dioxane solvent, whereas the sequence changed to B (4.01 kcal mol−1) → A (1.30 kcal mol−1) → C (1.15 kcal mol−1) in the nonpolar dichloromethane solvent. Additionally, it could be determined that the solvent polarity had a tremendous effect on compound A, whereas the effect on compound C was the smallest.
中文翻译:

不同的杂环和溶剂对三种新型光敏单甲酰化苯并恶唑衍生物的ESIPT机理的影响†
三种新型光活性单甲酰化苯并恶唑衍生物的荧光行为和特性A-C被发现由不同的杂环和溶剂的影响,如在最近的实验报告(Rodembusch,等人。,新J.化学。,2016年,40,2785)。不幸的是,这些化合物缺乏详细的激发态分子内质子转移(ESIPT)机制。在这项研究中,我们使用密度泛函理论(DFT)和时变DFT(TDDFT)方法来研究三种化合物A–C的动态ESIPT过程在两种不同的环境中(极性1,4-二恶烷和非极性二氯甲烷溶剂)。观察到的吸收光谱和荧光光谱与实验数据相互吻合,表明我们采用的TDDFT方法是可靠的。此外,通过分析两种溶剂中的键长,键角和IR振动光谱,可以确定这些化合物的分子内氢键(HBs)在S 1状态下得到增强,这可以促进ESIPT反应。此外,S 0和S 1之间的前沿分子轨道(MOs)和电子密度差的图状态显示分子内电荷转移,这为这三种化合物提供了ESIPT反应的可能性。此外,基于所构建的势能曲线,我们揭示了化合物A–C的详细动态ESIPT机理。结果,我们发现在极性1,4中,A(8.48 kcal mol -1)→ B(5.36 kcal mol -1)→ C(0.75 kcal mol -1)的化合物更有可能发生ESIPT过程-二恶烷溶剂,而顺序变为B(4.01 kcal mol -1)→ A(1.30 kcal mol -1)→ C(1.15 kcal mol -1)在非极性二氯甲烷溶剂中。此外,可以确定溶剂极性对化合物A的影响很大,而对化合物C的影响最小。
更新日期:2018-06-09
中文翻译:
不同的杂环和溶剂对三种新型光敏单甲酰化苯并恶唑衍生物的ESIPT机理的影响†
三种新型光活性单甲酰化苯并恶唑衍生物的荧光行为和特性A-C被发现由不同的杂环和溶剂的影响,如在最近的实验报告(Rodembusch,等人。,新J.化学。,2016年,40,2785)。不幸的是,这些化合物缺乏详细的激发态分子内质子转移(ESIPT)机制。在这项研究中,我们使用密度泛函理论(DFT)和时变DFT(TDDFT)方法来研究三种化合物A–C的动态ESIPT过程在两种不同的环境中(极性1,4-二恶烷和非极性二氯甲烷溶剂)。观察到的吸收光谱和荧光光谱与实验数据相互吻合,表明我们采用的TDDFT方法是可靠的。此外,通过分析两种溶剂中的键长,键角和IR振动光谱,可以确定这些化合物的分子内氢键(HBs)在S 1状态下得到增强,这可以促进ESIPT反应。此外,S 0和S 1之间的前沿分子轨道(MOs)和电子密度差的图状态显示分子内电荷转移,这为这三种化合物提供了ESIPT反应的可能性。此外,基于所构建的势能曲线,我们揭示了化合物A–C的详细动态ESIPT机理。结果,我们发现在极性1,4中,A(8.48 kcal mol -1)→ B(5.36 kcal mol -1)→ C(0.75 kcal mol -1)的化合物更有可能发生ESIPT过程-二恶烷溶剂,而顺序变为B(4.01 kcal mol -1)→ A(1.30 kcal mol -1)→ C(1.15 kcal mol -1)在非极性二氯甲烷溶剂中。此外,可以确定溶剂极性对化合物A的影响很大,而对化合物C的影响最小。


























 京公网安备 11010802027423号
京公网安备 11010802027423号