当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Dissociative Chemisorption of O2 on Al(111): Dynamics on a Correlated Wave-Function-Based Potential Energy Surface.
The Journal of Physical Chemistry Letters ( IF 5.7 ) Pub Date : 2018-06-05 , DOI: 10.1021/acs.jpclett.8b01470 Rongrong Yin 1 , Yaolong Zhang 1 , Florian Libisch 2 , Emily A Carter 3 , Hua Guo 4 , Bin Jiang 1
The Journal of Physical Chemistry Letters ( IF 5.7 ) Pub Date : 2018-06-05 , DOI: 10.1021/acs.jpclett.8b01470 Rongrong Yin 1 , Yaolong Zhang 1 , Florian Libisch 2 , Emily A Carter 3 , Hua Guo 4 , Bin Jiang 1
Affiliation
|
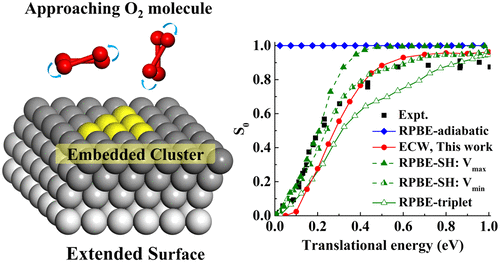
Dissociative chemisorption of O2 on the Al(111) surface represents an extensively studied prototype for understanding the interaction between O2 and metal surfaces. It is well known that the experimentally observed activation barrier for O2 dissociation is not captured by conventional density functional theory. The interpretation of this barrier as a result of spin transitions along the reaction path has been challenged by recent embedded correlated wave function (ECW) calculations that naturally yield an adiabatic barrier. However, the ECW calculations have been limited to a static analysis of the reaction pathways and have not yet been tested by dynamics simulations. We present a global six-dimensional potential energy surface (PES) for this system parametrized with ECW data points. This new PES provides a reasonable description of the site-specific and orientation-dependent activation barriers. Quasi-classical trajectory calculations on this PES semiquantitatively reproduce both the observed translational energy dependence of the sticking probability and steric effects with aligned O2 molecules.
中文翻译:

O2在Al(111)上的解离化学吸附:基于相关波函数的势能面的动力学。
O 2在Al(111)表面上的解离化学吸附代表了一个广泛研究的原型,用于理解O 2和金属表面之间的相互作用。众所周知,常规密度泛函理论并未捕获实验观察到的O2离解的活化势垒。由于最近沿嵌入的相关波函数(ECW)计算自然地产生了绝热势垒,因此对沿着反应路径自旋跃迁的结果对该势垒的解释提出了挑战。但是,ECW计算仅限于反应路径的静态分析,尚未通过动力学模拟进行测试。我们用ECW数据点为该系统提出了一个全球六维势能面(PES)。此新的PES提供了针对特定位置和依赖方向的激活障碍的合理描述。在该PES上的准经典轨迹计算半定量地再现了观察到的平移能量依赖性和排列的O2分子的粘着概率以及空间效应。
更新日期:2018-05-29
中文翻译:
O2在Al(111)上的解离化学吸附:基于相关波函数的势能面的动力学。
O 2在Al(111)表面上的解离化学吸附代表了一个广泛研究的原型,用于理解O 2和金属表面之间的相互作用。众所周知,常规密度泛函理论并未捕获实验观察到的O2离解的活化势垒。由于最近沿嵌入的相关波函数(ECW)计算自然地产生了绝热势垒,因此对沿着反应路径自旋跃迁的结果对该势垒的解释提出了挑战。但是,ECW计算仅限于反应路径的静态分析,尚未通过动力学模拟进行测试。我们用ECW数据点为该系统提出了一个全球六维势能面(PES)。此新的PES提供了针对特定位置和依赖方向的激活障碍的合理描述。在该PES上的准经典轨迹计算半定量地再现了观察到的平移能量依赖性和排列的O2分子的粘着概率以及空间效应。


























 京公网安备 11010802027423号
京公网安备 11010802027423号