当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Atomic Energies from a Convolutional Neural Network
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2018-05-29 00:00:00 , DOI: 10.1021/acs.jctc.8b00149 Xin Chen 1, 2 , Mathias S. Jørgensen 2 , Jun Li 1 , Bjørk Hammer 2
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2018-05-29 00:00:00 , DOI: 10.1021/acs.jctc.8b00149 Xin Chen 1, 2 , Mathias S. Jørgensen 2 , Jun Li 1 , Bjørk Hammer 2
Affiliation
|
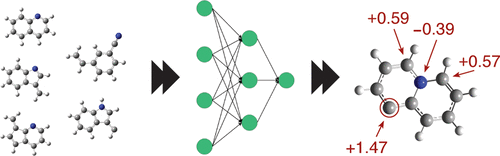
Understanding interactions and structural properties at the atomic level is often a prerequisite to the design of novel materials. Theoretical studies based on quantum-mechanical first-principles calculations can provide this knowledge but at an immense computational cost. In recent years, machine learning has been successful in predicting structural properties at a much lower cost. Here we propose a simplified structure descriptor with no empirical parameters, “k-Bags”, together with a scalable and comprehensive machine learning framework that can deepen our understanding of atomic properties of structures. This model can readily predict structure-energy relations that can provide results close to the accuracy of ab initio methods. The model provides chemically meaningful atomic energies enabling theoretical analysis of organic and inorganic molecular structures. Utilization of the local information provided by the atomic energies significantly improves upon the stochastic steps in our evolutionary global structure optimization, resulting in a much faster global minimum search of molecules, clusters, and surfaced supported species.
中文翻译:

卷积神经网络的原子能
在原子水平上理解相互作用和结构性质通常是新颖材料设计的先决条件。基于量子力学第一性原理计算的理论研究可以提供这一知识,但计算量巨大。近年来,机器学习已经以较低的成本成功地预测了结构特性。在这里,我们提出了一个没有经验参数的简化结构描述符“ k-Bags”,以及可扩展且全面的机器学习框架,该框架可以加深我们对结构原子性质的理解。该模型可以轻松预测结构-能量关系,从而可以提供接近于从头算的准确性的结果。该模型提供了具有化学意义的原子能,可以对有机和无机分子结构进行理论分析。利用原子能提供的局部信息可以显着改善我们进化全局结构优化中的随机步骤,从而更快地进行分子,团簇和表面受支持物种的全局最小搜索。
更新日期:2018-05-29
中文翻译:
卷积神经网络的原子能
在原子水平上理解相互作用和结构性质通常是新颖材料设计的先决条件。基于量子力学第一性原理计算的理论研究可以提供这一知识,但计算量巨大。近年来,机器学习已经以较低的成本成功地预测了结构特性。在这里,我们提出了一个没有经验参数的简化结构描述符“ k-Bags”,以及可扩展且全面的机器学习框架,该框架可以加深我们对结构原子性质的理解。该模型可以轻松预测结构-能量关系,从而可以提供接近于从头算的准确性的结果。该模型提供了具有化学意义的原子能,可以对有机和无机分子结构进行理论分析。利用原子能提供的局部信息可以显着改善我们进化全局结构优化中的随机步骤,从而更快地进行分子,团簇和表面受支持物种的全局最小搜索。


























 京公网安备 11010802027423号
京公网安备 11010802027423号