当前位置:
X-MOL 学术
›
Faraday Discuss.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
The importance of configurational disorder in crystal structure prediction: the case of loratadine
Faraday Discussions ( IF 3.4 ) Pub Date : 2018-05-15 , DOI: 10.1039/c8fd00072g Grahame R. Woollam 1, 2, 3 , Marcus A. Neumann 4, 5 , Trixie Wagner 2, 3, 6 , Roger J. Davey 7, 8, 9
Faraday Discussions ( IF 3.4 ) Pub Date : 2018-05-15 , DOI: 10.1039/c8fd00072g Grahame R. Woollam 1, 2, 3 , Marcus A. Neumann 4, 5 , Trixie Wagner 2, 3, 6 , Roger J. Davey 7, 8, 9
Affiliation
|
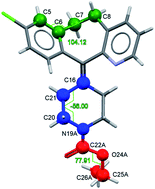
Loratadine, an over-the-counter antihistamine medication, has two known monotropically related polymorphs, both of which feature disorder. A combined experimental and computational approach using variable temperature single crystal X-ray diffraction (VT-SCXRD) analysis and dispersion corrected density functional theory (DFT-D) reveals that the nature of the disorder in each form is markedly different and cannot be described by a simple isolated-site model with thermally populated conformations in either of the two cases. In Form I, the ethyl carbamate functionality adopts two different configurations, with adjacent moieties interacting along one-dimensional chains. The most stable arrangement features alternating configurations, but because of the low energetic cost of stacking faults, the domain sizes are short and an average crystal structure is observed experimentally. The configurational free energy of the disordered structure is lower than the energy of the two corresponding ordered crystal structures, but the energy decrease is dominated by the lower lattice energy of the alternating arrangement with a small entropic contribution. In Form II, the flexible cycloheptane bridge adopts two different configurations. The disorder is not an equilibrium property but is instead frozen-in during the crystallisation process. The configurational free energy of the disordered structure falls in between the lattice energies of the two corresponding ordered structures. The two ordered components of each disordered structure are all found in a crystal structure prediction (CSP) study with the GRACE programme. However, the experimentally observed stability relationship is only reproduced when the energy contribution of disorder is taken into account. The disordered model of Form I is found to be lower in energy than all the other predicted structures and there is no indication of a missing, thermodynamically more stable, form. The case of loratadine demonstrates that experimentally observed disorder close to 50/50 does not necessarily correspond to a free energy decrease by kT ln 2.
中文翻译:

构型紊乱在晶体结构预测中的重要性:氯雷他定的案例
氯雷他定(Loratadine)是一种非处方抗组胺药,具有两种已知的与单向性相关的多晶型物,均具有疾病特征。使用可变温度单晶X射线衍射(VT-SCXRD)分析和色散校正的密度泛函理论(DFT-D)的组合实验和计算方法表明,每种形式的疾病的性质都明显不同,无法通过以下方式描述在这两种情况中的任何一种情况下,都具有带有热填充构象的简单隔离站点模型。在形式I中,氨基甲酸乙酯官能度采用两种不同的构型,相邻部分沿一维链相互作用。最稳定的布置具有交替配置,但是由于堆叠故障的能源成本较低,畴尺寸短,并且通过实验观察到平均晶体结构。无序结构的构型自由能低于两个相应的有序晶体结构的能,但是能量的下降主要是由交替排列的较低晶格能决定的,熵的贡献很小。在形式II中,柔性环庚烷桥采用两种不同的构型。无序不是平衡性质,而是在结晶过程中冻结。无序结构的构型自由能落在两个相应的有序结构的晶格能之间。每个无序结构的两个有序成分都可以通过GRACE程序在晶体结构预测(CSP)研究中找到。然而,只有在考虑到无序的能量贡献时,才能重现实验观察到的稳定性关系。发现形式I的无序模型的能量低于所有其他预测的结构,并且没有迹象表明缺少热力学上更稳定的形式。氯雷他定的案例表明,实验观察到的接近50/50的疾病不一定对应于自由能的降低kT ln 2。
更新日期:2018-10-26
中文翻译:
构型紊乱在晶体结构预测中的重要性:氯雷他定的案例
氯雷他定(Loratadine)是一种非处方抗组胺药,具有两种已知的与单向性相关的多晶型物,均具有疾病特征。使用可变温度单晶X射线衍射(VT-SCXRD)分析和色散校正的密度泛函理论(DFT-D)的组合实验和计算方法表明,每种形式的疾病的性质都明显不同,无法通过以下方式描述在这两种情况中的任何一种情况下,都具有带有热填充构象的简单隔离站点模型。在形式I中,氨基甲酸乙酯官能度采用两种不同的构型,相邻部分沿一维链相互作用。最稳定的布置具有交替配置,但是由于堆叠故障的能源成本较低,畴尺寸短,并且通过实验观察到平均晶体结构。无序结构的构型自由能低于两个相应的有序晶体结构的能,但是能量的下降主要是由交替排列的较低晶格能决定的,熵的贡献很小。在形式II中,柔性环庚烷桥采用两种不同的构型。无序不是平衡性质,而是在结晶过程中冻结。无序结构的构型自由能落在两个相应的有序结构的晶格能之间。每个无序结构的两个有序成分都可以通过GRACE程序在晶体结构预测(CSP)研究中找到。然而,只有在考虑到无序的能量贡献时,才能重现实验观察到的稳定性关系。发现形式I的无序模型的能量低于所有其他预测的结构,并且没有迹象表明缺少热力学上更稳定的形式。氯雷他定的案例表明,实验观察到的接近50/50的疾病不一定对应于自由能的降低kT ln 2。

























 京公网安备 11010802027423号
京公网安备 11010802027423号