当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
An Adaptive Pipeline To Maximize Isobaric Tagging Data in Large-Scale MS-Based Proteomics
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2018-05-04 , DOI: 10.1021/acs.jproteome.8b00110 John Corthésy 1 , Konstantinos Theofilatos 2 , Seferina Mavroudi 2, 3 , Charlotte Macron 1 , Ornella Cominetti 1 , Mona Remlawi 1 , Francesco Ferraro 1 , Antonio Núñez Galindo 1 , Martin Kussmann 1 , Spiridon Likothanassis 2, 4 , Loïc Dayon 1
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2018-05-04 , DOI: 10.1021/acs.jproteome.8b00110 John Corthésy 1 , Konstantinos Theofilatos 2 , Seferina Mavroudi 2, 3 , Charlotte Macron 1 , Ornella Cominetti 1 , Mona Remlawi 1 , Francesco Ferraro 1 , Antonio Núñez Galindo 1 , Martin Kussmann 1 , Spiridon Likothanassis 2, 4 , Loïc Dayon 1
Affiliation
|
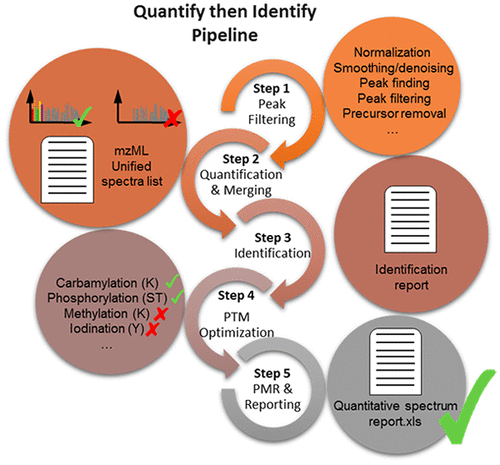
Isobaric tagging is the method of choice in mass-spectrometry-based proteomics for comparing several conditions at a time. Despite its multiplexing capabilities, some drawbacks appear when multiple experiments are merged for comparison in large sample-size studies due to the presence of missing values, which result from the stochastic nature of the data-dependent acquisition mode. Another indirect cause of data incompleteness might derive from the proteomic-typical data-processing workflow that first identifies proteins in individual experiments and then only quantifies those identified proteins, leaving a large number of unmatched spectra with quantitative information unexploited. Inspired by untargeted metabolomic and label-free proteomic workflows, we developed a quantification-driven bioinformatic pipeline (Quantify then Identify (QtI)) that optimizes the processing of isobaric tandem mass tag (TMT) data from large-scale studies. This pipeline includes innovative features, such as peak filtering with a self-adaptive preprocessing pipeline optimization method, Peptide Match Rescue, and Optimized Post-Translational Modification. QtI outperforms a classical benchmark workflow in terms of quantification and identification rates, significantly reducing missing data while preserving unmatched features for quantitative comparison. The number of unexploited tandem mass spectra was reduced by 77 and 62% for two human cerebrospinal fluid and plasma data sets, respectively.
中文翻译:

大规模基于MS的蛋白质组学中最大化等压标记数据的自适应管道
等压标记是在基于质谱的蛋白质组学中一次比较多个条件的选择方法。尽管它具有多路复用功能,但由于存在缺失值(由于数据相关的采集模式的随机性质而导致的缺失),当将多个实验合并用于大样本量研究中进行比较时,仍会出现一些缺陷。数据不完整的另一个间接原因可能来自蛋白质组学典型的数据处理工作流程,该工作流程首先在单个实验中鉴定蛋白质,然后仅对鉴定出的蛋白质进行定量,从而留下大量无法利用的定量信息而无法利用的光谱。受非靶向代谢组学和无标签蛋白质组学工作流程的启发,我们开发了量化驱动的生物信息流水线(先量化再识别(QtI)),以优化大规模研究中的等压串联质量标签(TMT)数据的处理。该流水线包括创新功能,例如采用自适应预处理流水线优化方法的峰过滤,肽段匹配救援和优化的翻译后修饰。在定量和鉴定率方面,QtI优于传统的基准工作流程,可显着减少丢失的数据,同时保留无与伦比的功能以进行定量比较。对于两个人的脑脊液和血浆数据集,未利用的串联质谱数分别减少了77%和62%。该流水线包括创新功能,例如采用自适应预处理流水线优化方法的峰过滤,肽段匹配救援和优化的翻译后修饰。在定量和鉴定率方面,QtI优于传统的基准工作流程,可显着减少丢失的数据,同时保留无与伦比的功能以进行定量比较。对于两个人的脑脊液和血浆数据集,未利用的串联质谱数分别减少了77%和62%。该流水线包括创新功能,例如采用自适应预处理流水线优化方法的峰过滤,肽段匹配救援和优化的翻译后修饰。在定量和鉴定率方面,QtI优于传统的基准工作流程,可显着减少丢失的数据,同时保留无与伦比的功能以进行定量比较。对于两个人的脑脊液和血浆数据集,未利用的串联质谱数分别减少了77%和62%。大大减少了丢失的数据,同时保留了无与伦比的功能以进行定量比较。对于两个人脑脊髓液和血浆数据集,未利用的串联质谱数分别减少了77%和62%。大大减少了丢失的数据,同时保留了无与伦比的功能以进行定量比较。对于两个人的脑脊液和血浆数据集,未利用的串联质谱数分别减少了77%和62%。
更新日期:2018-05-04
中文翻译:
大规模基于MS的蛋白质组学中最大化等压标记数据的自适应管道
等压标记是在基于质谱的蛋白质组学中一次比较多个条件的选择方法。尽管它具有多路复用功能,但由于存在缺失值(由于数据相关的采集模式的随机性质而导致的缺失),当将多个实验合并用于大样本量研究中进行比较时,仍会出现一些缺陷。数据不完整的另一个间接原因可能来自蛋白质组学典型的数据处理工作流程,该工作流程首先在单个实验中鉴定蛋白质,然后仅对鉴定出的蛋白质进行定量,从而留下大量无法利用的定量信息而无法利用的光谱。受非靶向代谢组学和无标签蛋白质组学工作流程的启发,我们开发了量化驱动的生物信息流水线(先量化再识别(QtI)),以优化大规模研究中的等压串联质量标签(TMT)数据的处理。该流水线包括创新功能,例如采用自适应预处理流水线优化方法的峰过滤,肽段匹配救援和优化的翻译后修饰。在定量和鉴定率方面,QtI优于传统的基准工作流程,可显着减少丢失的数据,同时保留无与伦比的功能以进行定量比较。对于两个人的脑脊液和血浆数据集,未利用的串联质谱数分别减少了77%和62%。该流水线包括创新功能,例如采用自适应预处理流水线优化方法的峰过滤,肽段匹配救援和优化的翻译后修饰。在定量和鉴定率方面,QtI优于传统的基准工作流程,可显着减少丢失的数据,同时保留无与伦比的功能以进行定量比较。对于两个人的脑脊液和血浆数据集,未利用的串联质谱数分别减少了77%和62%。该流水线包括创新功能,例如采用自适应预处理流水线优化方法的峰过滤,肽段匹配救援和优化的翻译后修饰。在定量和鉴定率方面,QtI优于传统的基准工作流程,可显着减少丢失的数据,同时保留无与伦比的功能以进行定量比较。对于两个人的脑脊液和血浆数据集,未利用的串联质谱数分别减少了77%和62%。大大减少了丢失的数据,同时保留了无与伦比的功能以进行定量比较。对于两个人脑脊髓液和血浆数据集,未利用的串联质谱数分别减少了77%和62%。大大减少了丢失的数据,同时保留了无与伦比的功能以进行定量比较。对于两个人的脑脊液和血浆数据集,未利用的串联质谱数分别减少了77%和62%。


























 京公网安备 11010802027423号
京公网安备 11010802027423号