当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Plasticity of the Binding Site of Renin: Optimized Selection of Protein Structures for Ensemble Docking
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-04-23 00:00:00 , DOI: 10.1021/acs.jcim.8b00010 Claas Strecker 1 , Bernd Meyer 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-04-23 00:00:00 , DOI: 10.1021/acs.jcim.8b00010 Claas Strecker 1 , Bernd Meyer 1
Affiliation
|
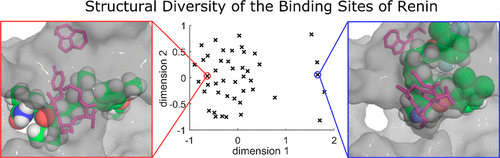
Protein flexibility poses a major challenge to docking of potential ligands in that the binding site can adopt different shapes. Docking algorithms usually keep the protein rigid and only allow the ligand to be treated as flexible. However, a wrong assessment of the shape of the binding pocket can prevent a ligand from adapting a correct pose. Ensemble docking is a simple yet promising method to solve this problem: Ligands are docked into multiple structures, and the results are subsequently merged. Selection of protein structures is a significant factor for this approach. In this work we perform a comprehensive and comparative study evaluating the impact of structure selection on ensemble docking. We perform ensemble docking with several crystal structures and with structures derived from molecular dynamics simulations of renin, an attractive target for antihypertensive drugs. Here, 500 ns of MD simulations revealed binding site shapes not found in any available crystal structure. We evaluate the importance of structure selection for ensemble docking by comparing binding pose prediction, ability to rank actives above nonactives (screening utility), and scoring accuracy. As a result, for ensemble definition k-means clustering appears to be better suited than hierarchical clustering with average linkage. The best performing ensemble consists of four crystal structures and is able to reproduce the native ligand poses better than any individual crystal structure. Moreover this ensemble outperforms 88% of all individual crystal structures in terms of screening utility as well as scoring accuracy. Similarly, ensembles of MD-derived structures perform on average better than 75% of any individual crystal structure in terms of scoring accuracy at all inspected ensembles sizes.
中文翻译:

肾素结合位点的可塑性:集合坞的蛋白质结构的优化选择。
蛋白质的柔韧性对潜在配体的对接构成了重大挑战,因为结合位点可以采用不同的形状。对接算法通常使蛋白质保持刚性,仅允许将配体视为柔性的。但是,对结合袋形状的错误评估会阻止配体适应正确的姿势。集成对接是解决此问题的一种简单而有希望的方法:将配体对接成多个结构,然后合并结果。蛋白质结构的选择是这种方法的重要因素。在这项工作中,我们进行了全面的比较研究,评估了结构选择对整体对接的影响。我们通过几个晶体结构以及肾素分子动力学模拟得出的结构进行整体对接,降压药的一个有吸引力的目标。在这里,500 ns的MD模拟显示在任何可用的晶体结构中都找不到结合位点形状。我们通过比较结合姿势预测,将非活性物质以上的活性物质进行排序的能力(筛选实用程序)和评分准确度,来评估结构选择对整体对接的重要性。结果,用于整体定义k均值聚类似乎比具有平均链接的层次聚类更适合。表现最佳的合奏由四个晶体结构组成,并且能够比任何单个晶体结构更好地再现天然配体姿势。此外,在筛选实用性和评分准确性方面,该整体表现优于所有单个晶体结构的88%。类似地,就所有检查的集成块尺寸而言,在评分准确性方面,MD派生的集成块的平均表现要好于任何单个晶体结构的75%。
更新日期:2018-04-23
中文翻译:
肾素结合位点的可塑性:集合坞的蛋白质结构的优化选择。
蛋白质的柔韧性对潜在配体的对接构成了重大挑战,因为结合位点可以采用不同的形状。对接算法通常使蛋白质保持刚性,仅允许将配体视为柔性的。但是,对结合袋形状的错误评估会阻止配体适应正确的姿势。集成对接是解决此问题的一种简单而有希望的方法:将配体对接成多个结构,然后合并结果。蛋白质结构的选择是这种方法的重要因素。在这项工作中,我们进行了全面的比较研究,评估了结构选择对整体对接的影响。我们通过几个晶体结构以及肾素分子动力学模拟得出的结构进行整体对接,降压药的一个有吸引力的目标。在这里,500 ns的MD模拟显示在任何可用的晶体结构中都找不到结合位点形状。我们通过比较结合姿势预测,将非活性物质以上的活性物质进行排序的能力(筛选实用程序)和评分准确度,来评估结构选择对整体对接的重要性。结果,用于整体定义k均值聚类似乎比具有平均链接的层次聚类更适合。表现最佳的合奏由四个晶体结构组成,并且能够比任何单个晶体结构更好地再现天然配体姿势。此外,在筛选实用性和评分准确性方面,该整体表现优于所有单个晶体结构的88%。类似地,就所有检查的集成块尺寸而言,在评分准确性方面,MD派生的集成块的平均表现要好于任何单个晶体结构的75%。


























 京公网安备 11010802027423号
京公网安备 11010802027423号