当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Ground-State Charge-Density Distribution in a Crystal of the Luminescent ortho-Phenylenediboronic Acid Complex with 8-Hydroxyquinoline
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2018-04-19 00:00:00 , DOI: 10.1021/acs.jpca.8b00832 Katarzyna N. Jarzembska 1 , Radosław Kamiński 1 , Krzysztof Durka 2 , Krzysztof Woźniak 3
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2018-04-19 00:00:00 , DOI: 10.1021/acs.jpca.8b00832 Katarzyna N. Jarzembska 1 , Radosław Kamiński 1 , Krzysztof Durka 2 , Krzysztof Woźniak 3
Affiliation
|
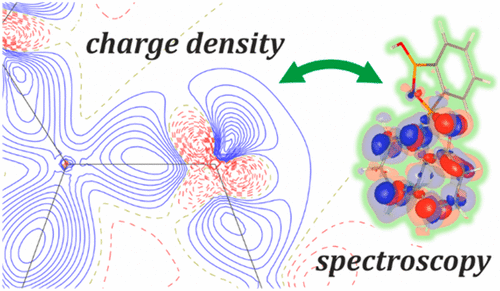
This contribution is devoted to the first electron density studies of a luminescent oxyquinolinato boron complex in the solid state. ortho-Phenylenediboronic acid mixed with 8-hydroxyquinoline in dioxane forms high-quality single crystals via slow solvent evaporation, which allows successful high resolution data collection (sin θ/λ = 1.2 Å–1) and charge density distribution modeling. Particular attention has been paid to the boron–oxygen fragment connecting the two parts of the complex, and to the solvent species exhibiting anharmonic thermal motion. The experiment and theory compared rather well in terms of atomic charges and volumes, except for the boron centers. Boron atoms, as expected, constitute the most electron-deficient species in the complex molecule, whereas the neighboring oxygen and carbon atoms are the most significantly negatively charged ones. This part of the molecule appears to be very much involved in the charge transfer occurring between the acid fragment and oxyquinoline moiety leading to the observed fluorescence, as supported by the time-dependent density functional theory (TDDFT) results and the generated transition density maps. TDDFT calculations indicated that p-type atomic orbitals contributing to the HOMO–1, HOMO, and LUMO play the major role in the lowest energy transitions, and enabled further comparison with the charge density features, which is discussed in details. Furthermore, the results confirmed the known fact the Q ligand character is most important for the spectroscopic properties of this class of complexes.
中文翻译:

发光的邻-苯二硼酸与8-羟基喹啉配合物晶体中的基态电荷密度分布
该贡献致力于固态的发光氧基喹啉基硼配合物的首次电子密度研究。在二恶烷中将邻苯二硼酸与8-羟基喹啉混合,通过缓慢的溶剂蒸发形成高质量的单晶,从而可以成功地进行高分辨率数据收集(sinθ/λ= 1.2Å –1)和电荷密度分布建模。特别注意连接复合物两个部分的硼-氧片段,以及表现出非谐热运动的溶剂。实验和理论在原子电荷和体积方面进行了很好的比较,除了硼中心。正如预期的那样,硼原子是络合物分子中最缺乏电子的物种,而相邻的氧和碳原子则是带负电荷最明显的物种。分子的这一部分似乎非常参与酸片段和羟基喹啉部分之间发生的电荷转移,从而导致观察到的荧光,这是由时间依赖性密度泛函理论(TDDFT)结果和生成的跃迁密度图所支持的。促成HOMO-1,HOMO和LUMO的p型原子轨道在最低的能量跃迁中起主要作用,并使得能够与电荷密度特征作进一步的比较,这将在后面详细讨论。此外,结果证实了已知的事实,即Q配体特征对于这类配合物的光谱性质是最重要的。
更新日期:2018-04-19
中文翻译:
发光的邻-苯二硼酸与8-羟基喹啉配合物晶体中的基态电荷密度分布
该贡献致力于固态的发光氧基喹啉基硼配合物的首次电子密度研究。在二恶烷中将邻苯二硼酸与8-羟基喹啉混合,通过缓慢的溶剂蒸发形成高质量的单晶,从而可以成功地进行高分辨率数据收集(sinθ/λ= 1.2Å –1)和电荷密度分布建模。特别注意连接复合物两个部分的硼-氧片段,以及表现出非谐热运动的溶剂。实验和理论在原子电荷和体积方面进行了很好的比较,除了硼中心。正如预期的那样,硼原子是络合物分子中最缺乏电子的物种,而相邻的氧和碳原子则是带负电荷最明显的物种。分子的这一部分似乎非常参与酸片段和羟基喹啉部分之间发生的电荷转移,从而导致观察到的荧光,这是由时间依赖性密度泛函理论(TDDFT)结果和生成的跃迁密度图所支持的。促成HOMO-1,HOMO和LUMO的p型原子轨道在最低的能量跃迁中起主要作用,并使得能够与电荷密度特征作进一步的比较,这将在后面详细讨论。此外,结果证实了已知的事实,即Q配体特征对于这类配合物的光谱性质是最重要的。


























 京公网安备 11010802027423号
京公网安备 11010802027423号