当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Probing Charge Delocalization in Solid State Polychromophoric Cation Radicals Using X-ray Crystallography and DFT Calculations
The Journal of Physical Chemistry C ( IF 3.7 ) Pub Date : 2018-04-13 00:00:00 , DOI: 10.1021/acs.jpcc.8b02184 Lena V. Ivanova 1 , Denan Wang 1 , Sergey Lindeman 1 , Maxim V. Ivanov 1 , Rajendra Rathore 1
The Journal of Physical Chemistry C ( IF 3.7 ) Pub Date : 2018-04-13 00:00:00 , DOI: 10.1021/acs.jpcc.8b02184 Lena V. Ivanova 1 , Denan Wang 1 , Sergey Lindeman 1 , Maxim V. Ivanov 1 , Rajendra Rathore 1
Affiliation
|
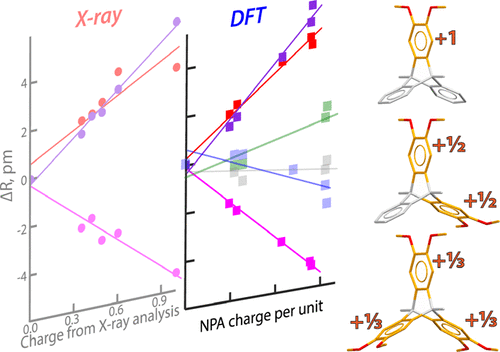
Assessing the charge delocalization in polychromophoric assemblies is a critical step toward designing novel charge transfer materials. Triptycene-based materials are particularly attractive, owing to their unique packing arrangement in the solid state. Here, we systematically probe, both experimentally (with X-ray crystallography) and theoretically (using Density Functional Theory, DFT), the extent of cationic charge (i.e., hole) delocalization in a set of triptycene derivatives with one, two, and three electron-rich 1,2-dimethoxybenzenoid (veratrole) rings. We demonstrate that the amount of charge at each veratrole can be deduced from experiment by analysis of the oxidation-induced bond length changes in comparison with a model compound containing one veratrole ring as a reference. In contrast, DFT calculations provide not only oxidation-induced structural reorganization, but also the charge distribution with the aid of natural population analysis. A comparative analysis shows that both experiment and theory are of equal efficacy in quantifying the extent of hole distribution in polychromophoric cation radicals, despite issues of packing, solvent molecules, and counterions that are present in the crystals. Therefore, combining X-ray crystallographic data with insight from DFT calculations can provide a detailed understanding of the hole distribution in polychromophoric cation radicals, in turn allowing an informed design of the next-generation charge-transport materials based on triptycene and other polychromophoric scaffolds.
中文翻译:

使用X射线晶体学和DFT计算探测固态多色阳离子自由基中的电荷离域
评估多发色组装体中的电荷离域化是设计新型电荷转移材料的关键一步。基于三萜烯的材料由于其独特的固态堆积布置而特别具有吸引力。在这里,我们通过实验(使用X射线晶体学)和理论上(使用密度泛函理论,DFT)系统地研究一组具有一个,两个和三个三茂金属衍生物的阳离子电荷(即空穴)离域化的程度富电子的1,2-二甲氧基苯甲环(veratrole)。我们证明,与包含一个Veratrole环作为参考的模型化合物相比,通过分析氧化诱导的键长变化可以从实验中推导每个Veratrole的电荷量。相比之下,DFT计算不仅提供了氧化诱导的结构重组,而且还借助自然种群分析提供了电荷分布。对比分析表明,尽管存在晶体中存在堆积,溶剂分子和抗衡离子的问题,实验和理论在量化多发色阳离子自由基中空穴分布的程度方面均具有相同的功效。因此,将X射线晶体学数据与DFT计算的洞察力相结合,可以提供对多发色性阳离子自由基中空穴分布的详细了解,从而可以基于三萜烯和其他多发色性支架,对下一代电荷传输材料进行明智的设计。对比分析表明,尽管存在晶体中存在堆积,溶剂分子和抗衡离子的问题,实验和理论在量化多发色阳离子自由基中空穴分布的程度方面均具有相同的功效。因此,将X射线晶体学数据与DFT计算的洞察力相结合,可以提供对多发色性阳离子自由基中空穴分布的详细了解,从而可以基于三萜烯和其他多发色性支架,对下一代电荷传输材料进行明智的设计。对比分析表明,尽管存在晶体中存在堆积,溶剂分子和抗衡离子的问题,但实验和理论在量化多发色阳离子自由基中空穴分布的程度方面均具有相同的功效。因此,将X射线晶体学数据与DFT计算的洞察力相结合,可以提供对多发色性阳离子自由基中空穴分布的详细了解,从而可以基于三萜烯和其他多发色性支架,对下一代电荷传输材料进行明智的设计。
更新日期:2018-04-13
中文翻译:
使用X射线晶体学和DFT计算探测固态多色阳离子自由基中的电荷离域
评估多发色组装体中的电荷离域化是设计新型电荷转移材料的关键一步。基于三萜烯的材料由于其独特的固态堆积布置而特别具有吸引力。在这里,我们通过实验(使用X射线晶体学)和理论上(使用密度泛函理论,DFT)系统地研究一组具有一个,两个和三个三茂金属衍生物的阳离子电荷(即空穴)离域化的程度富电子的1,2-二甲氧基苯甲环(veratrole)。我们证明,与包含一个Veratrole环作为参考的模型化合物相比,通过分析氧化诱导的键长变化可以从实验中推导每个Veratrole的电荷量。相比之下,DFT计算不仅提供了氧化诱导的结构重组,而且还借助自然种群分析提供了电荷分布。对比分析表明,尽管存在晶体中存在堆积,溶剂分子和抗衡离子的问题,实验和理论在量化多发色阳离子自由基中空穴分布的程度方面均具有相同的功效。因此,将X射线晶体学数据与DFT计算的洞察力相结合,可以提供对多发色性阳离子自由基中空穴分布的详细了解,从而可以基于三萜烯和其他多发色性支架,对下一代电荷传输材料进行明智的设计。对比分析表明,尽管存在晶体中存在堆积,溶剂分子和抗衡离子的问题,实验和理论在量化多发色阳离子自由基中空穴分布的程度方面均具有相同的功效。因此,将X射线晶体学数据与DFT计算的洞察力相结合,可以提供对多发色性阳离子自由基中空穴分布的详细了解,从而可以基于三萜烯和其他多发色性支架,对下一代电荷传输材料进行明智的设计。对比分析表明,尽管存在晶体中存在堆积,溶剂分子和抗衡离子的问题,但实验和理论在量化多发色阳离子自由基中空穴分布的程度方面均具有相同的功效。因此,将X射线晶体学数据与DFT计算的洞察力相结合,可以提供对多发色性阳离子自由基中空穴分布的详细了解,从而可以基于三萜烯和其他多发色性支架,对下一代电荷传输材料进行明智的设计。

























 京公网安备 11010802027423号
京公网安备 11010802027423号