当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
A Protein Standard That Emulates Homology for the Characterization of Protein Inference Algorithms
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2018-04-16 , DOI: 10.1021/acs.jproteome.7b00899 Matthew The 1 , Fredrik Edfors 1 , Yasset Perez-Riverol 2 , Samuel H. Payne 3 , Michael R. Hoopmann 4 , Magnus Palmblad 5 , Björn Forsström 1 , Lukas Käll 1
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2018-04-16 , DOI: 10.1021/acs.jproteome.7b00899 Matthew The 1 , Fredrik Edfors 1 , Yasset Perez-Riverol 2 , Samuel H. Payne 3 , Michael R. Hoopmann 4 , Magnus Palmblad 5 , Björn Forsström 1 , Lukas Käll 1
Affiliation
|
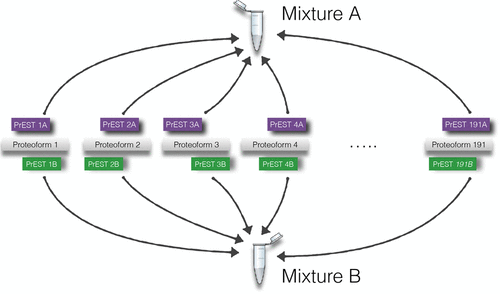
A natural way to benchmark the performance of an analytical experimental setup is to use samples of known composition and see to what degree one can correctly infer the content of such a sample from the data. For shotgun proteomics, one of the inherent problems of interpreting data is that the measured analytes are peptides and not the actual proteins themselves. As some proteins share proteolytic peptides, there might be more than one possible causative set of proteins resulting in a given set of peptides and there is a need for mechanisms that infer proteins from lists of detected peptides. A weakness of commercially available samples of known content is that they consist of proteins that are deliberately selected for producing tryptic peptides that are unique to a single protein. Unfortunately, such samples do not expose any complications in protein inference. Hence, for a realistic benchmark of protein inference procedures, there is a need for samples of known content where the present proteins share peptides with known absent proteins. Here, we present such a standard, that is based on E. coli expressed human protein fragments. To illustrate the application of this standard, we benchmark a set of different protein inference procedures on the data. We observe that inference procedures excluding shared peptides provide more accurate estimates of errors compared to methods that include information from shared peptides, while still giving a reasonable performance in terms of the number of identified proteins. We also demonstrate that using a sample of known protein content without proteins with shared tryptic peptides can give a false sense of accuracy for many protein inference methods.
中文翻译:

一种模拟同源性的蛋白质标准,用于蛋白质推理算法的表征
基准分析实验装置性能的一种自然方法是使用已知成分的样品,并查看可以从数据中正确推断出此类样品的含量的程度。对于shot弹枪蛋白质组学来说,解释数据的固有问题之一是所测分析物是肽,而不是实际的蛋白质本身。由于某些蛋白质共享蛋白水解肽,因此可能会有不止一种可能的致病蛋白质组产生给定的一组肽,因此需要从检测到的肽列表中推断蛋白质的机制。已知含量的可商购样品的一个弱点是它们由故意选择的蛋白质组成,以产生单一蛋白质特有的胰蛋白酶肽。很遗憾,这样的样品不会在蛋白质推断中暴露出任何复杂性。因此,对于蛋白质推断程序的现实基准,需要具有已知含量的样品,其中本发明的蛋白质与已知的缺乏蛋白质共享肽。在这里,我们提出了这样一个标准,它基于大肠杆菌表达了人类蛋白质片段。为了说明该标准的应用,我们在数据上对一组不同的蛋白质推断程序进行了基准测试。我们观察到,与包括共享肽段信息的方法相比,排除共享肽段的推理程序可提供更准确的错误估计,同时在鉴定蛋白质的数量方面仍能提供合理的性能。我们还证明,使用没有共享胰蛋白酶肽的蛋白质的已知蛋白质含量的样品可能会给许多蛋白质推断方法带来错误的准确性。
更新日期:2018-04-17
中文翻译:
一种模拟同源性的蛋白质标准,用于蛋白质推理算法的表征
基准分析实验装置性能的一种自然方法是使用已知成分的样品,并查看可以从数据中正确推断出此类样品的含量的程度。对于shot弹枪蛋白质组学来说,解释数据的固有问题之一是所测分析物是肽,而不是实际的蛋白质本身。由于某些蛋白质共享蛋白水解肽,因此可能会有不止一种可能的致病蛋白质组产生给定的一组肽,因此需要从检测到的肽列表中推断蛋白质的机制。已知含量的可商购样品的一个弱点是它们由故意选择的蛋白质组成,以产生单一蛋白质特有的胰蛋白酶肽。很遗憾,这样的样品不会在蛋白质推断中暴露出任何复杂性。因此,对于蛋白质推断程序的现实基准,需要具有已知含量的样品,其中本发明的蛋白质与已知的缺乏蛋白质共享肽。在这里,我们提出了这样一个标准,它基于大肠杆菌表达了人类蛋白质片段。为了说明该标准的应用,我们在数据上对一组不同的蛋白质推断程序进行了基准测试。我们观察到,与包括共享肽段信息的方法相比,排除共享肽段的推理程序可提供更准确的错误估计,同时在鉴定蛋白质的数量方面仍能提供合理的性能。我们还证明,使用没有共享胰蛋白酶肽的蛋白质的已知蛋白质含量的样品可能会给许多蛋白质推断方法带来错误的准确性。

























 京公网安备 11010802027423号
京公网安备 11010802027423号