当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Orotidine 5'-Monophosphate Decarboxylase: Probing the Limits of the Possible for Enzyme Catalysis.
Accounts of Chemical Research ( IF 18.3 ) Pub Date : 2018-03-29 , DOI: 10.1021/acs.accounts.8b00059 John P Richard 1 , Tina L Amyes 1 , Archie C Reyes 1
Accounts of Chemical Research ( IF 18.3 ) Pub Date : 2018-03-29 , DOI: 10.1021/acs.accounts.8b00059 John P Richard 1 , Tina L Amyes 1 , Archie C Reyes 1
Affiliation
|
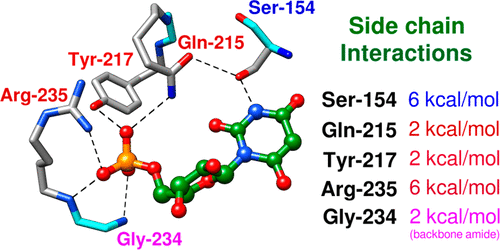
The mystery associated with catalysis by what were once regarded as protein black boxes, diminished with the X-ray crystallographic determination of the three-dimensional structures of enzyme-substrate complexes. The report that several high-resolution X-ray crystal structures of orotidine 5'-monophosphate decarboxylase (OMPDC) failed to provide a consensus mechanism for enzyme-catalyzed decarboxylation of OMP to form uridine 5'-monophosphate, therefore, provoked a flurry of controversy. This controversy was fueled by the enormous 1023-fold rate acceleration for this enzyme, which had " jolted many biochemists' assumptions about the catalytic potential of enzymes." Our studies on the mechanism of action of OMPDC provide strong evidence that catalysis by this enzyme is not fundamentally different from less proficient catalysts, while highlighting important architectural elements that enable a peak level of performance. Many enzymes undergo substrate-induced protein conformational changes that trap their substrates in solvent occluded protein cages, but the conformational change induced by ligand binding to OMPDC is incredibly complex, as required to enable the development of 22 kcal/mol of stabilizing binding interactions with the phosphodianion and ribosyl substrate fragments of OMP. The binding energy from these fragments is utilized to activate OMPDC for catalysis of decarboxylation at the orotate fragment of OMP, through the creation of a tight, catalytically active, protein cage from the floppy, open, unliganded form of OMPDC. Such utilization of binding energy for ligand-driven conformational changes provides a general mechanism to obtain specificity in transition state binding. The rate enhancement that results from the binding of carbon acid substrates to enzymes is partly due to a reduction in the carbon acid p Ka that is associated with ligand binding. The binding of UMP to OMPDC results in an unusually large >12 unit decrease in the p Ka = 29 for abstraction of the C-6 substrate hydrogen, due to stabilization of an enzyme-bound vinyl carbanion, which is also an intermediate of OMPDC-catalyzed decarboxylation. The protein-ligand interactions operate to stabilize the vinyl carbanion at the enzyme active site compared to aqueous solution, rather than to stabilize the transition state for the concerted electrophilic displacement of CO2 by H+ that avoids formation of this reaction intermediate. There is evidence that OMPDC induces strain into the bound substrate. The interaction between the amide side chain of Gln-215 from the phosphodianion gripper loop and the hydroxymethylene side chain of Ser-154 from the pyrimidine umbrella of ScOMPDC position the amide side chain to interact with the phosphodianion of OMP. There are no direct stabilizing interactions between dianion gripper protein side chains Gln-215, Tyr-217, and Arg-235 and the pyrimidine ring at the decarboxylation transition state. Rather these side chains function solely to hold OMPDC in the catalytically active closed conformation. The hydrophobic side chains that line the active site of OMPDC in the region of the departing CO2 product may function to stabilize the decarboxylation transition state by providing hydrophobic solvation of this product.
中文翻译:

奥罗替丁 5'-单磷酸脱羧酶:探索酶催化的可能性极限。
曾经被认为是蛋白质黑匣子的催化作用的神秘面纱,随着对酶-底物复合物三维结构的 X 射线晶体学测定而消失。乳清酸5'-单磷酸脱羧酶(OMPDC)的几种高分辨率X射线晶体结构的报道未能为OMP的酶催化脱羧形成尿苷5'-单磷酸提供共识机制,因此引发了一阵争论. 这种酶以 1023 倍的巨大速度加速引发了这场争议,这“颠覆了许多生物化学家关于酶催化潜力的假设”。我们对 OMPDC 作用机制的研究提供了强有力的证据,表明这种酶的催化作用与效率较低的催化剂没有根本区别,同时突出显示可实现最高性能水平的重要架构元素。许多酶经历底物诱导的蛋白质构象变化,将它们的底物捕获在溶剂封闭的蛋白质笼中,但配体与 OMPDC 结合诱导的构象变化非常复杂,这是开发 22 kcal/mol 稳定结合相互作用所必需的。 OMP 的磷酸二价阴离子和核糖基底物片段。来自这些片段的结合能用于激活 OMPDC,以通过从松散、开放、未配体形式的 OMPDC 中创建紧密、催化活性的蛋白质笼来催化 OMP 的乳清酸盐片段处的脱羧作用。这种将结合能用于配体驱动的构象变化提供了获得过渡态结合特异性的一般机制。由碳酸底物与酶结合引起的速率提高部分是由于与配体结合相关的碳酸p Ka 的降低。由于酶结合的乙烯基碳负离子(也是 OMPDC 的中间体)的稳定性,UMP 与 OMPDC 的结合导致用于提取 C-6 底物氢的 p Ka = 29 出现异常大的 >12 个单位降低催化脱羧。与水溶液相比,蛋白质-配体相互作用可稳定酶活性位点处的乙烯基碳负离子,而不是通过 H+ 稳定 CO2 协同亲电置换的过渡态,从而避免形成该反应中间体。有证据表明 OMPDC 将应变引入结合的底物。来自磷酸二阴离子夹持环的 Gln-215 的酰胺侧链与来自 ScOMPDC 嘧啶伞的 Ser-154 的羟基亚甲基侧链之间的相互作用使酰胺侧链与 OMP 的磷酸二阴离子相互作用。二价阴离子夹持蛋白侧链 Gln-215、Tyr-217 和 Arg-235 与脱羧过渡态的嘧啶环之间没有直接的稳定相互作用。相反,这些侧链仅用于将 OMPDC 保持在具有催化活性的闭合构象中。
更新日期:2018-03-29
中文翻译:
奥罗替丁 5'-单磷酸脱羧酶:探索酶催化的可能性极限。
曾经被认为是蛋白质黑匣子的催化作用的神秘面纱,随着对酶-底物复合物三维结构的 X 射线晶体学测定而消失。乳清酸5'-单磷酸脱羧酶(OMPDC)的几种高分辨率X射线晶体结构的报道未能为OMP的酶催化脱羧形成尿苷5'-单磷酸提供共识机制,因此引发了一阵争论. 这种酶以 1023 倍的巨大速度加速引发了这场争议,这“颠覆了许多生物化学家关于酶催化潜力的假设”。我们对 OMPDC 作用机制的研究提供了强有力的证据,表明这种酶的催化作用与效率较低的催化剂没有根本区别,同时突出显示可实现最高性能水平的重要架构元素。许多酶经历底物诱导的蛋白质构象变化,将它们的底物捕获在溶剂封闭的蛋白质笼中,但配体与 OMPDC 结合诱导的构象变化非常复杂,这是开发 22 kcal/mol 稳定结合相互作用所必需的。 OMP 的磷酸二价阴离子和核糖基底物片段。来自这些片段的结合能用于激活 OMPDC,以通过从松散、开放、未配体形式的 OMPDC 中创建紧密、催化活性的蛋白质笼来催化 OMP 的乳清酸盐片段处的脱羧作用。这种将结合能用于配体驱动的构象变化提供了获得过渡态结合特异性的一般机制。由碳酸底物与酶结合引起的速率提高部分是由于与配体结合相关的碳酸p Ka 的降低。由于酶结合的乙烯基碳负离子(也是 OMPDC 的中间体)的稳定性,UMP 与 OMPDC 的结合导致用于提取 C-6 底物氢的 p Ka = 29 出现异常大的 >12 个单位降低催化脱羧。与水溶液相比,蛋白质-配体相互作用可稳定酶活性位点处的乙烯基碳负离子,而不是通过 H+ 稳定 CO2 协同亲电置换的过渡态,从而避免形成该反应中间体。有证据表明 OMPDC 将应变引入结合的底物。来自磷酸二阴离子夹持环的 Gln-215 的酰胺侧链与来自 ScOMPDC 嘧啶伞的 Ser-154 的羟基亚甲基侧链之间的相互作用使酰胺侧链与 OMP 的磷酸二阴离子相互作用。二价阴离子夹持蛋白侧链 Gln-215、Tyr-217 和 Arg-235 与脱羧过渡态的嘧啶环之间没有直接的稳定相互作用。相反,这些侧链仅用于将 OMPDC 保持在具有催化活性的闭合构象中。


























 京公网安备 11010802027423号
京公网安备 11010802027423号