Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Planning chemical syntheses with deep neural networks and symbolic AI
Nature ( IF 64.8 ) Pub Date : 2018-03-01 , DOI: 10.1038/nature25978 Marwin H. S. Segler , Mike Preuss , Mark P. Waller
Nature ( IF 64.8 ) Pub Date : 2018-03-01 , DOI: 10.1038/nature25978 Marwin H. S. Segler , Mike Preuss , Mark P. Waller
|
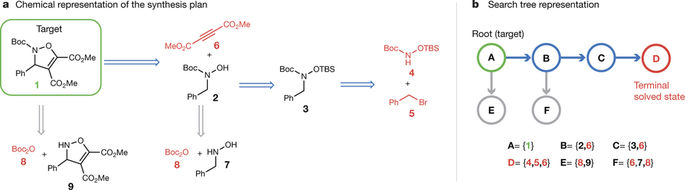
From medicines to materials, small organic molecules are indispensable for human well-being. To plan their syntheses, chemists employ a problem solving technique called retrosynthesis. In retrosynthesis, target molecules are recursively transformed into increasingly simpler precursor compounds until a set of readily available starting materials is obtained. Computer-aided retrosynthesis would be a highly valuable tool, however, past approaches were slow and provided results of unsatisfactory quality. Here, we employ Monte Carlo Tree Search (MCTS) to efficiently discover retrosynthetic routes. MCTS was combined with an expansion policy network that guides the search, and an “in-scope” filter network to pre-select the most promising retrosynthetic steps. These deep neural networks were trained on 12 million reactions, which represents essentially all reactions ever published in organic chemistry. Our system solves almost twice as many molecules and is 30 times faster in comparison to the traditional search method based on extracted rules and hand-coded heuristics. Finally after a 60 year history of computer-aided synthesis planning, chemists can no longer distinguish between routes generated by a computer system and real routes taken from the scientific literature. We anticipate that our method will accelerate drug and materials discovery by assisting chemists to plan better syntheses faster, and by enabling fully automated robot synthesis. Retrosynthetic analysis is the canonical technique to plan the synthesis of organic small molecules, for example drugs, agroand fine chemicals, and is part of every chemist’s curriculum.[1, 2] In retrosynthesis, a search tree is built by “working-backwards”, analysing molecules recursively and transforming them into simpler precursors until one obtains a set of known or commercially available building block molecules.[3] The transformations are reversed chemical reactions: A chemist can take the plan, and execute it in the lab in the forward direction to synthesize the target compound. Retrosynthetic analysis is a formidable intellectual exercise that demands broad and deep chemical knowledge. Its central element requires a combination of creativity and a pattern recognition process, in which functional groups (patterns of atoms and bonds) of the target molecule are matched to transformations.[4] Transformations are derived from successfully conducted series of similar reactions with analogous starting materials, and often named after their discoverers.[5] At each retrosynthetic step, out of thousands of known transformations in modern chemistry, chemists intuitively rank the most promising transformations highly, and do not even actively think about the unreasonable ones. Unfortunately, when a transform is applied to a new molecule, there is no guarantee that it will generalize and the corresponding reaction will actually proceed in the expected way.[7] A molecule failing to react in the way predicted by the transform is called ’out of scope’. This can be due to steric or electronic effects, an incomplete understanding of the reaction mechanism, or conflicting reactivity in In cognitive science, this can is explained via “System 1” and “System 2” in dual process theory.[6] the molecular context, which may hamper a reaction and lead to chemo-, regioor stereoselectivity issues. Predicting which molecules are ’within scope’ can be challenging even for the best human chemists, who perform synthesis planning using knowledge and mechanistic reasoning acquired during long years of study and the many intuitive models the chemical community developed over the last 250 years, in combination with extensive literature research for every transform step, which may take hours.[7, 3] Computer-assisted synthesis planning (CASP) could help chemists to find better routes faster, and is a missing component in virtual de novo design and robot systems performing molecular designsynthesis-test cycles.[8–11] To perform CASP, the knowledge that humans “simply learn” has to be transferred into an executable program.[12, 4, 13–15] Despite 60 years of research and industrialscale backing, attempts to formalize chemistry by manual encoding by experts have not convinced synthetic chemists.[15–20] Approaches to algorithmically extract transformations from reaction datasets[21–23] on a purely symbolic level were criticized for their high amount of noise and lack of “chemical intelligence”.[14, 13] However, we recently showed that deep neural networks trained on large reaction datasets are capable of ranking extracted symbolic transformations, and can learn to avoid reactivity conflicts.[24] This allows to mimic the expert’s intuitive decision-making.[24] b) Search Tree Representation a) Chemical Representation of the Synthesis Plan
中文翻译:

使用深度神经网络和符号 AI 规划化学合成
从药物到材料,有机小分子对人类福祉不可或缺。为了计划他们的合成,化学家采用了一种称为逆合成的问题解决技术。在逆合成中,目标分子被递归地转化为越来越简单的前体化合物,直到获得一组容易获得的起始材料。计算机辅助的逆合成将是一种非常有价值的工具,但是,过去的方法速度缓慢并且提供的结果质量不令人满意。在这里,我们采用蒙特卡罗树搜索 (MCTS) 来有效地发现逆合成路线。MCTS 与指导搜索的扩展策略网络和“范围内”过滤网络相结合,以预先选择最有希望的逆合成步骤。这些深度神经网络接受了 1200 万次反应的训练,它基本上代表了有机化学中曾经发表过的所有反应。与基于提取规则和手工编码启发式的传统搜索方法相比,我们的系统解决了几乎两倍的分子,速度提高了 30 倍。最后,经过 60 年的计算机辅助合成规划历史,化学家再也无法区分计算机系统生成的路线和科学文献中的真实路线。我们预计,我们的方法将通过协助化学家更快地规划更好的合成,并通过实现全自动机器人合成来加速药物和材料的发现。逆合成分析是规划有机小分子(例如药物、农业和精细化学品)合成的经典技术,是每位化学家课程的一部分。 [1, 2] 在逆合成中,搜索树是通过“向后工作”构建的,递归地分析分子并将它们转化为更简单的前体,直到获得一组已知的或商业上可用的构建块分子。 [3] 转化是逆向化学反应:化学家可以制定计划,并在实验室中正向执行以合成目标化合物。逆合成分析是一项艰巨的智力活动,需要广泛而深入的化学知识。其核心要素需要创造力和模式识别过程的结合,其中目标分子的官能团(原子和键的模式)与转换相匹配。 [4] 转化来源于使用类似起始材料成功进行的一系列类似反应,并且通常以其发现者的名字命名。[5] 在每一个逆合成步骤中,在现代化学中已知的数千种转化中,化学家凭直觉对最有希望的转化进行高度评价,甚至不会主动考虑不合理的转化。不幸的是,当变换应用于新分子时,不能保证它会泛化并且相应的反应实际上会以预期的方式进行。 [7] 未能以转换预测的方式反应的分子被称为“超出范围”。这可能是由于空间或电子效应,对反应机制的不完全理解,或在认知科学中相互矛盾的反应性,这可以通过双重过程理论中的“系统 1”和“系统 2”来解释。 [6] 分子环境,这可能会阻碍反应并导致化学、区域或立体选择性问题。通过专家手动编码将化学形式化的尝试并没有说服合成化学家。 [15-20] 在纯符号级别上从反应数据集 [21-23] 中通过算法提取转换的方法因其大量的噪音和缺乏“化学智能”。[14, 13] 然而,我们最近表明,在大型反应数据集上训练的深度神经网络能够对提取的符号转换进行排序,并且可以学习避免反应性冲突。[24] 这允许模仿专家的直觉决策。 [24] b) 搜索树表示 a) 合成计划的化学表示 [15-20] 在纯符号层面从反应数据集 [21-23] 中通过算法提取变换的方法因其大量噪声和缺乏“化学智能”而受到批评。 [14, 13] 然而,我们最近表明在大型反应数据集上训练的深度神经网络能够对提取的符号转换进行排序,并且可以学习避免反应性冲突。 [24] 这允许模仿专家的直觉决策。 [24] b) 搜索树表示 a) 合成计划的化学表示 [15-20] 在纯符号层面从反应数据集 [21-23] 中通过算法提取变换的方法因其大量噪声和缺乏“化学智能”而受到批评。 [14, 13] 然而,我们最近表明在大型反应数据集上训练的深度神经网络能够对提取的符号转换进行排序,并且可以学习避免反应性冲突。 [24] 这允许模仿专家的直觉决策。 [24] b) 搜索树表示 a) 合成计划的化学表示
更新日期:2018-03-01
中文翻译:
使用深度神经网络和符号 AI 规划化学合成
从药物到材料,有机小分子对人类福祉不可或缺。为了计划他们的合成,化学家采用了一种称为逆合成的问题解决技术。在逆合成中,目标分子被递归地转化为越来越简单的前体化合物,直到获得一组容易获得的起始材料。计算机辅助的逆合成将是一种非常有价值的工具,但是,过去的方法速度缓慢并且提供的结果质量不令人满意。在这里,我们采用蒙特卡罗树搜索 (MCTS) 来有效地发现逆合成路线。MCTS 与指导搜索的扩展策略网络和“范围内”过滤网络相结合,以预先选择最有希望的逆合成步骤。这些深度神经网络接受了 1200 万次反应的训练,它基本上代表了有机化学中曾经发表过的所有反应。与基于提取规则和手工编码启发式的传统搜索方法相比,我们的系统解决了几乎两倍的分子,速度提高了 30 倍。最后,经过 60 年的计算机辅助合成规划历史,化学家再也无法区分计算机系统生成的路线和科学文献中的真实路线。我们预计,我们的方法将通过协助化学家更快地规划更好的合成,并通过实现全自动机器人合成来加速药物和材料的发现。逆合成分析是规划有机小分子(例如药物、农业和精细化学品)合成的经典技术,是每位化学家课程的一部分。 [1, 2] 在逆合成中,搜索树是通过“向后工作”构建的,递归地分析分子并将它们转化为更简单的前体,直到获得一组已知的或商业上可用的构建块分子。 [3] 转化是逆向化学反应:化学家可以制定计划,并在实验室中正向执行以合成目标化合物。逆合成分析是一项艰巨的智力活动,需要广泛而深入的化学知识。其核心要素需要创造力和模式识别过程的结合,其中目标分子的官能团(原子和键的模式)与转换相匹配。 [4] 转化来源于使用类似起始材料成功进行的一系列类似反应,并且通常以其发现者的名字命名。[5] 在每一个逆合成步骤中,在现代化学中已知的数千种转化中,化学家凭直觉对最有希望的转化进行高度评价,甚至不会主动考虑不合理的转化。不幸的是,当变换应用于新分子时,不能保证它会泛化并且相应的反应实际上会以预期的方式进行。 [7] 未能以转换预测的方式反应的分子被称为“超出范围”。这可能是由于空间或电子效应,对反应机制的不完全理解,或在认知科学中相互矛盾的反应性,这可以通过双重过程理论中的“系统 1”和“系统 2”来解释。 [6] 分子环境,这可能会阻碍反应并导致化学、区域或立体选择性问题。通过专家手动编码将化学形式化的尝试并没有说服合成化学家。 [15-20] 在纯符号级别上从反应数据集 [21-23] 中通过算法提取转换的方法因其大量的噪音和缺乏“化学智能”。[14, 13] 然而,我们最近表明,在大型反应数据集上训练的深度神经网络能够对提取的符号转换进行排序,并且可以学习避免反应性冲突。[24] 这允许模仿专家的直觉决策。 [24] b) 搜索树表示 a) 合成计划的化学表示 [15-20] 在纯符号层面从反应数据集 [21-23] 中通过算法提取变换的方法因其大量噪声和缺乏“化学智能”而受到批评。 [14, 13] 然而,我们最近表明在大型反应数据集上训练的深度神经网络能够对提取的符号转换进行排序,并且可以学习避免反应性冲突。 [24] 这允许模仿专家的直觉决策。 [24] b) 搜索树表示 a) 合成计划的化学表示 [15-20] 在纯符号层面从反应数据集 [21-23] 中通过算法提取变换的方法因其大量噪声和缺乏“化学智能”而受到批评。 [14, 13] 然而,我们最近表明在大型反应数据集上训练的深度神经网络能够对提取的符号转换进行排序,并且可以学习避免反应性冲突。 [24] 这允许模仿专家的直觉决策。 [24] b) 搜索树表示 a) 合成计划的化学表示
























 京公网安备 11010802027423号
京公网安备 11010802027423号