当前位置:
X-MOL 学术
›
Acta Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Ab initio magnesium-solute transport database using exact diffusion theory
Acta Materialia ( IF 9.4 ) Pub Date : 2018-05-01 , DOI: 10.1016/j.actamat.2018.03.025 Ravi Agarwal , Dallas R. Trinkle
Acta Materialia ( IF 9.4 ) Pub Date : 2018-05-01 , DOI: 10.1016/j.actamat.2018.03.025 Ravi Agarwal , Dallas R. Trinkle
|
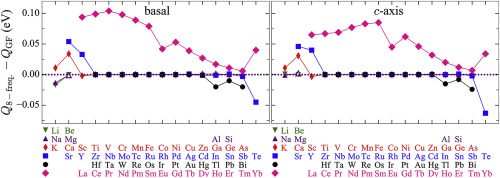
Abstract A recently developed Green function approach informed by ab initio calculations models vacancy-mediated transport of 61 solutes in a hexagonal close packed magnesium. The 8- and 13-frequency diffusion models approximate vacancy jump rates near a solute, leading to the inaccurate calculation of Onsager coefficients. We identify all the symmetry-unique vacancy jumps in the Mg lattice and use the Green function approach to calculate the Onsager coefficients exactly in the limit of dilute solute concentration. Density functional theory-computed solute-vacancy interactions and vacancy jump rates inform the Green function approach and previous diffusion models. Solutes with positive size misfit diffuse faster compared to the self-diffusion of Mg due to the relaxation of solute towards vacancy while solutes with negative size misfit diffuse slower. Transition metal solutes show drag for attractive solute-vacancy binding as well as for repulsive binding, due to faster reorientation rates of the vacancy around the solute compared to dissociation rates. Solutes from the s-block, p-block and lanthanide series with attractive solute-vacancy binding and slower reorientation rates compared to the dissociation rates show drag due to vacancy motion around the solute through alternate dissociation and association jumps. The prediction of activation energy of diffusion from the 8-frequency model deviates by more than 50 meV for solutes with significant correlations effect. Our GF approach prediction of solute diffusion coefficients agree well with the available experimental measurements.
中文翻译:

使用精确扩散理论的从头算镁溶质传输数据库
摘要 最近开发的格林函数方法由 ab initio 计算得出,模拟了六方密堆积镁中 61 种溶质的空位介导传输。8 和 13 频率扩散模型近似于溶质附近的空位跳跃率,导致 Onsager 系数的计算不准确。我们确定了 Mg 晶格中所有对称唯一的空位跳跃,并使用格林函数方法在稀溶质浓度极限内精确计算 Onsager 系数。密度泛函理论计算的溶质空位相互作用和空位跳跃率告知格林函数方法和以前的扩散模型。由于溶质向空位松弛,与 Mg 的自扩散相比,具有正尺寸失配的溶质扩散得更快,而具有负尺寸失配的溶质扩散更慢。由于与解离速率相比,溶质周围空位的重新定向速率更快,因此过渡金属溶质对有吸引力的溶质-空位结合以及排斥结合都具有阻力。来自 s-block、p-block 和镧系元素的溶质与解离速率相比具有有吸引力的溶质空位结合和较慢的重新定向速率,显示出由于围绕溶质的空位运动通过交替解离和结合跳跃而产生的阻力。对于具有显着相关效应的溶质,8 频率模型对扩散活化能的预测偏差超过 50 meV。
更新日期:2018-05-01
中文翻译:
使用精确扩散理论的从头算镁溶质传输数据库
摘要 最近开发的格林函数方法由 ab initio 计算得出,模拟了六方密堆积镁中 61 种溶质的空位介导传输。8 和 13 频率扩散模型近似于溶质附近的空位跳跃率,导致 Onsager 系数的计算不准确。我们确定了 Mg 晶格中所有对称唯一的空位跳跃,并使用格林函数方法在稀溶质浓度极限内精确计算 Onsager 系数。密度泛函理论计算的溶质空位相互作用和空位跳跃率告知格林函数方法和以前的扩散模型。由于溶质向空位松弛,与 Mg 的自扩散相比,具有正尺寸失配的溶质扩散得更快,而具有负尺寸失配的溶质扩散更慢。由于与解离速率相比,溶质周围空位的重新定向速率更快,因此过渡金属溶质对有吸引力的溶质-空位结合以及排斥结合都具有阻力。来自 s-block、p-block 和镧系元素的溶质与解离速率相比具有有吸引力的溶质空位结合和较慢的重新定向速率,显示出由于围绕溶质的空位运动通过交替解离和结合跳跃而产生的阻力。对于具有显着相关效应的溶质,8 频率模型对扩散活化能的预测偏差超过 50 meV。

























 京公网安备 11010802027423号
京公网安备 11010802027423号