European Journal of Medicinal Chemistry ( IF 6.7 ) Pub Date : 2018-03-16 , DOI: 10.1016/j.ejmech.2018.03.040 Agnese Chiara Pippione , Irene Maria Carnovale , Davide Bonanni , Marcella Sini , Parveen Goyal , Elisabetta Marini , Klaus Pors , Salvatore Adinolfi , Daniele Zonari , Claudio Festuccia , Weixiao Yuan Wahlgren , Rosmarie Friemann , Renzo Bagnati , Donatella Boschi , Simonetta Oliaro-Bosso , Marco Lucio Lolli
|
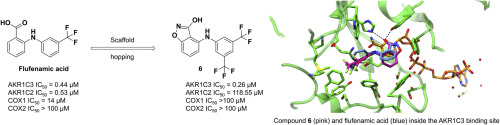
The aldo-keto reductase 1C3 (AKR1C3) isoform plays a vital role in the biosynthesis of androgens and is considered an attractive target in prostate cancer (PCa). No AKR1C3-targeted agent has to date been approved for clinical use. Flufenamic acid and indomethacine are non-steroidal anti-inflammatory drugs known to inhibit AKR1C3 in a non-selective manner as COX off-target effects are also observed. Recently, we employed a scaffold hopping approach to design a new class of potent and selective AKR1C3 inhibitors based on a N-substituted hydroxylated triazole pharmacophore. Following a similar strategy, we designed a new series focused around an acidic hydroxybenzoisoxazole moiety, which was rationalised to mimic the benzoic acid role in the flufenamic scaffold. Through iterative rounds of drug design, synthesis and biological evaluation, several compounds were discovered to target AKR1C3 in a selective manner. The most promising compound of series (6) was found to be highly selective (up to 450-fold) for AKR1C3 over the 1C2 isoform with minimal COX1 and COX2 off-target effects. Other inhibitors were obtained modulating the best example of hydroxylated triazoles we previously presented. In cell-based assays, the most promising compounds of both series reduced the cell proliferation, prostate specific antigen (PSA) and testosterone production in AKR1C3-expressing 22RV1 prostate cancer cells and showed synergistic effect when assayed in combination with abiraterone and enzalutamide. Structure determination of AKR1C3 co-crystallized with one representative compound from each of the two series clearly identified both compounds in the androstenedione binding site, hence supporting the biochemical data.
中文翻译:
基于苯并异恶唑部分的强效选择性醛酮还原酶1C3(AKR1C3)抑制剂:生物等排骨架跳跃法在氟苯那酸中的应用
醛酮还原酶1C3(AKR1C3)亚型在雄激素的生物合成中起着至关重要的作用,被认为是前列腺癌(PCa)的诱人靶标。迄今为止,尚未批准将AKR1C3靶向药物用于临床。氟苯那酸和吲哚美辛是非甾体类抗炎药,已知还会以非选择性方式抑制AKR1C3,因为还观察到了COX脱靶效应。最近,我们采用脚手架跳跃方法来设计基于N的新型有效和选择性的AKR1C3抑制剂取代的羟基化三唑药效基团。遵循类似的策略,我们设计了一个围绕酸性羟基苯并异恶唑部分的新系列,该部分被合理化以模仿苯甲酸在氟芬那支架中的作用。通过反复的药物设计,合成和生物学评估,发现了几种化合物以选择性方式靶向AKR1C3。系列中最有前途的化合物(6)被发现对AKR1C3的选择性比对1C2异构体高(高达450倍),而COX1和COX2脱靶效应最小。获得了其他抑制剂,这些抑制剂调节了我们先前介绍的羟基化三唑的最佳实例。在基于细胞的分析中,两个系列中最有前途的化合物均降低了表达AKR1C3的22RV1前列腺癌细胞的细胞增殖,前列腺特异性抗原(PSA)和睾丸激素的产生,与阿比特龙和恩杂鲁胺联合检测时显示出协同作用。与一种代表性化合物共结晶的AKR1C3的结构测定,可以清楚地从雄烯二酮结合位点鉴定出这两种化合物,从而支持了生化数据。

























 京公网安备 11010802027423号
京公网安备 11010802027423号