当前位置:
X-MOL 学术
›
Mol. Syst. Des. Eng.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
COSMO-CAMPD: a framework for integrated design of molecules and processes based on COSMO-RS†
Molecular Systems Design & Engineering ( IF 3.6 ) Pub Date : 2018-03-16 00:00:00 , DOI: 10.1039/c7me00125h J. Scheffczyk 1, 2, 3, 4 , P. Schäfer 2, 3, 4, 5 , L. Fleitmann 1, 2, 3, 4 , J. Thien 1, 2, 3, 4 , C. Redepenning 2, 3, 4, 5 , K. Leonhard 1, 2, 3, 4 , W. Marquardt 2, 3, 4, 5 , A. Bardow 1, 2, 3, 4, 6
Molecular Systems Design & Engineering ( IF 3.6 ) Pub Date : 2018-03-16 00:00:00 , DOI: 10.1039/c7me00125h J. Scheffczyk 1, 2, 3, 4 , P. Schäfer 2, 3, 4, 5 , L. Fleitmann 1, 2, 3, 4 , J. Thien 1, 2, 3, 4 , C. Redepenning 2, 3, 4, 5 , K. Leonhard 1, 2, 3, 4 , W. Marquardt 2, 3, 4, 5 , A. Bardow 1, 2, 3, 4, 6
Affiliation
|
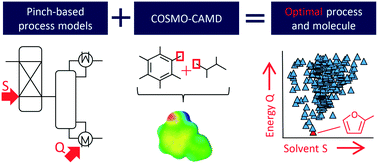
Molecules are often the key for energetically and economically efficient processes and thus need to be carefully selected. The simultaneous optimization of molecules and processes is addressed by computer-aided molecular and process design (CAMPD). Thereby, the design problem captures directly the complex interaction of molecules and processes. The success of CAMPD strongly relies on the available model to predict the molecular properties. Typically, simplified thermodynamic prediction methods such as group contribution (GC) methods are used. GC methods assume the additivity of functional groups and require interaction parameters determined in experiments which limits the molecular design space. In this work, we overcome these limitations by presenting COSMO-CAMPD, a framework for integrated design of molecules and processes based on COSMO-RS. COSMO-RS employs quantum-mechanics (QM)-based thermodynamic property predictions and thus does not rely on experimental data or group additivity. We embed the COSMO-RS predictions in an efficient QM calculation strategy using two COSMO-RS accuracy levels. This strategy reduces computational cost to less than 1 minute per molecule. Designed molecules are evaluated at the process level using highly efficient and thermodynamically accurate pinch-based process models. With the results from pinch-based process models, rigorous process flowsheet simulations are initialized and easily converged in a subsequent refinement step using Aspen Plus. We apply the COSMO-CAMPD framework to a case study for solvent design in a hybrid extraction–distillation process for the bio-based platform chemical γ-valerolactone. COSMO-CAMPD successfully designs molecules as solvents for an optimized process which reduces the energy demand by 63% in comparison to published literature solvents. The predictions of COSMO-CAMPD are challenged by liquid–liquid equilibrium experiments for the top designed commercially available solvents 2-methylfuran and toluene. For the most promising commercially available solvent with practical relevance, 2-methylfuran, the experiments lead to 50% reduction in process energy demand compared to the benchmark solvent n-butyl acetate corresponding well to the reduction of 47% predicted by COSMO-CAMPD. The experimental validation highlights the practical value of COSMO-CAMPD.
中文翻译:

COSMO-CAMPD:基于COSMO-RS的分子和过程集成设计的框架†
分子通常是在能源和经济上高效的过程的关键,因此需要仔细选择。分子和过程的同时优化通过计算机辅助分子和过程设计(CAMPD)解决。因此,设计问题直接捕获了分子与过程的复杂相互作用。CAMPD的成功在很大程度上取决于可用的模型来预测分子特性。通常,使用简化的热力学预测方法,例如基团贡献(GC)方法。GC方法假定官能团具有可加性,并且需要在实验中确定相互作用参数,从而限制了分子设计空间。在这项工作中,我们通过提出COSMO-CAMPD克服了这些限制,COSMO-CAMPD是基于COSMO-RS的分子和过程集成设计的框架。COSMO-RS采用基于量子力学(QM)的热力学性质预测,因此不依赖于实验数据或基团可加性。我们使用两个COSMO-RS精度级别将COSMO-RS预测嵌入到有效的QM计算策略中。该策略可将计算成本降低到每个分子少于1分钟。使用高效且热力学精确的基于捏的过程模型在过程级别评估设计的分子。利用基于捏合过程模型的结果,可以对严格的过程流程图进行初始化,并在随后的精制步骤中使用Aspen Plus轻松将其收敛。我们将COSMO-CAMPD框架应用于基于生物的平台化学γ-戊内酯的混合萃取-蒸馏过程中溶剂设计的案例研究。COSMO-CAMPD成功地将分子设计为溶剂,用于优化工艺,与已发表的文献溶剂相比,该技术将能源需求降低了63%。COSMO-CAMPD的预测受到液-液平衡实验的挑战,该实验针对的是顶级设计的可商购获得的溶剂2-甲基呋喃和甲苯。对于具有实用意义的最有希望的可商购获得的溶剂2-甲基呋喃,与基准溶剂相比,该实验可将过程能源需求减少50%乙酸正丁酯与COSMO-CAMPD预测的减少47%很好。实验验证突出了COSMO-CAMPD的实用价值。
更新日期:2018-03-16
中文翻译:
COSMO-CAMPD:基于COSMO-RS的分子和过程集成设计的框架†
分子通常是在能源和经济上高效的过程的关键,因此需要仔细选择。分子和过程的同时优化通过计算机辅助分子和过程设计(CAMPD)解决。因此,设计问题直接捕获了分子与过程的复杂相互作用。CAMPD的成功在很大程度上取决于可用的模型来预测分子特性。通常,使用简化的热力学预测方法,例如基团贡献(GC)方法。GC方法假定官能团具有可加性,并且需要在实验中确定相互作用参数,从而限制了分子设计空间。在这项工作中,我们通过提出COSMO-CAMPD克服了这些限制,COSMO-CAMPD是基于COSMO-RS的分子和过程集成设计的框架。COSMO-RS采用基于量子力学(QM)的热力学性质预测,因此不依赖于实验数据或基团可加性。我们使用两个COSMO-RS精度级别将COSMO-RS预测嵌入到有效的QM计算策略中。该策略可将计算成本降低到每个分子少于1分钟。使用高效且热力学精确的基于捏的过程模型在过程级别评估设计的分子。利用基于捏合过程模型的结果,可以对严格的过程流程图进行初始化,并在随后的精制步骤中使用Aspen Plus轻松将其收敛。我们将COSMO-CAMPD框架应用于基于生物的平台化学γ-戊内酯的混合萃取-蒸馏过程中溶剂设计的案例研究。COSMO-CAMPD成功地将分子设计为溶剂,用于优化工艺,与已发表的文献溶剂相比,该技术将能源需求降低了63%。COSMO-CAMPD的预测受到液-液平衡实验的挑战,该实验针对的是顶级设计的可商购获得的溶剂2-甲基呋喃和甲苯。对于具有实用意义的最有希望的可商购获得的溶剂2-甲基呋喃,与基准溶剂相比,该实验可将过程能源需求减少50%乙酸正丁酯与COSMO-CAMPD预测的减少47%很好。实验验证突出了COSMO-CAMPD的实用价值。

























 京公网安备 11010802027423号
京公网安备 11010802027423号