当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Dispersion interactions between neighboring Bi atoms in (BiH3 )2 and Te(BiR2 )2
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-03-13 , DOI: 10.1002/jcc.25209 Rebekka Haack 1 , Stephan Schulz 1 , Georg Jansen 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-03-13 , DOI: 10.1002/jcc.25209 Rebekka Haack 1 , Stephan Schulz 1 , Georg Jansen 1
Affiliation
|
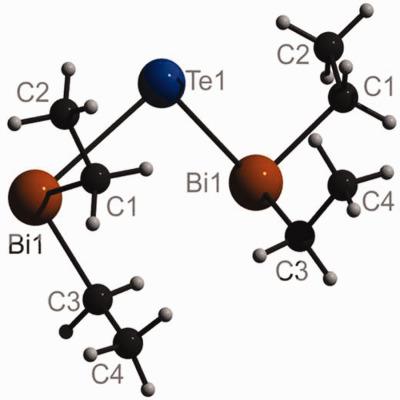
Triggered by the observation of a short Bi⋯Bi distance and a BiTeBi bond angle of only 86.6° in the crystal structure of bis(diethylbismuthanyl)tellurane quantum chemical computations on interactions between neighboring Bi atoms in Te(BiR2)2 molecules (R = H, Me, Et) and in (BiH3)2 were undertaken. Bi⋯Bi distances atoms were found to significantly shorten upon inclusion of the d shells of the heavy metal atoms into the electron correlation treatment, and it was confirmed that interaction energies from spin component‐scaled second‐order Møller–Plesset theory (SCS‐MP2) agree well with coupled‐cluster singles and doubles theory including perturbative triples (CCSD(T)). Density functional theory‐based symmetry‐adapted perturbation theory (DFT‐SAPT) was used to study the anisotropy of the interplay of dispersion attraction and steric repulsion between the Bi atoms. Finally, geometries and relative stabilities of syn–syn and syn–anti conformers of Te(BiR2)2 (R = H, Me, Et) and interconversion barriers between them were computed. © 2018 Wiley Periodicals, Inc.
中文翻译:

(BiH3 )2 和 Te(BiR2 )2 中相邻 Bi 原子之间的色散相互作用
在双(二乙基铋)碲烷的晶体结构中观察到短的 Bi⋯Bi 距离和仅 86.6° 的 BiTeBi 键角触发了 Te(BiR2)2 分子中相邻 Bi 原子之间相互作用的量子化学计算(R = H, Me, Et) 和 (BiH3)2 中进行。在将重金属原子的 d 壳层纳入电子关联处理后,发现 Bi⋯Bi 距离原子显着缩短,并且证实了来自自旋分量缩放的二阶 Møller-Plesset 理论(SCS-MP2 ) 与包括微扰三元组 (CCSD(T)) 的耦合簇单打和双打理论一致。基于密度泛函理论的对称性适应微扰理论 (DFT-SAPT) 用于研究 Bi 原子之间色散吸引力和空间排斥相互作用的各向异性。最后,计算Te(BiR2)2 (R = H, Me, Et) 的syn-syn 和syn-anti 构象异构体的几何形状和相对稳定性以及它们之间的相互转换势垒。© 2018 Wiley Periodicals, Inc.
更新日期:2018-03-13
中文翻译:
(BiH3 )2 和 Te(BiR2 )2 中相邻 Bi 原子之间的色散相互作用
在双(二乙基铋)碲烷的晶体结构中观察到短的 Bi⋯Bi 距离和仅 86.6° 的 BiTeBi 键角触发了 Te(BiR2)2 分子中相邻 Bi 原子之间相互作用的量子化学计算(R = H, Me, Et) 和 (BiH3)2 中进行。在将重金属原子的 d 壳层纳入电子关联处理后,发现 Bi⋯Bi 距离原子显着缩短,并且证实了来自自旋分量缩放的二阶 Møller-Plesset 理论(SCS-MP2 ) 与包括微扰三元组 (CCSD(T)) 的耦合簇单打和双打理论一致。基于密度泛函理论的对称性适应微扰理论 (DFT-SAPT) 用于研究 Bi 原子之间色散吸引力和空间排斥相互作用的各向异性。最后,计算Te(BiR2)2 (R = H, Me, Et) 的syn-syn 和syn-anti 构象异构体的几何形状和相对稳定性以及它们之间的相互转换势垒。© 2018 Wiley Periodicals, Inc.


























 京公网安备 11010802027423号
京公网安备 11010802027423号