当前位置:
X-MOL 学术
›
Organometallics
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Rhodium-Catalyzed Hydrocarboxylation: Mechanistic Analysis Reveals Unusual Transition State for Carbon–Carbon Bond Formation
Organometallics ( IF 2.8 ) Pub Date : 2018-03-12 00:00:00 , DOI: 10.1021/acs.organomet.7b00899 Ljiljana Pavlovic 1 , Janakiram Vaitla 2 , Annette Bayer 2 , Kathrin H. Hopmann 1
Organometallics ( IF 2.8 ) Pub Date : 2018-03-12 00:00:00 , DOI: 10.1021/acs.organomet.7b00899 Ljiljana Pavlovic 1 , Janakiram Vaitla 2 , Annette Bayer 2 , Kathrin H. Hopmann 1
Affiliation
|
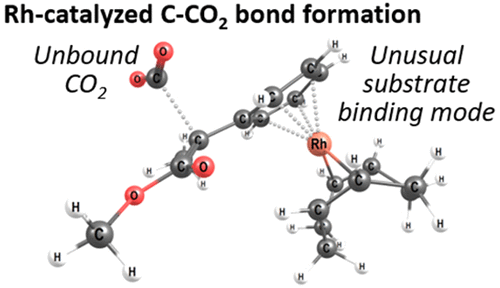
The mechanism of rhodium-COD-catalyzed hydrocarboxylation of styrene derivatives and α,β-unsaturated carbonyl compounds with CO2 has been investigated using density functional theory (PBE-D2/IEFPCM). The calculations support a catalytic cycle as originally proposed by Mikami and co-workers including β-hydride elimination, insertion of the unsaturated substrate into a rhodium–hydride bond, and subsequent carboxylation with CO2. The CO2 insertion step is found to be rate limiting. The calculations reveal two interesting aspects. First, during C–CO2 bond formation, the CO2 molecule interacts with neither the rhodium complex nor the organozinc additive. This appears to be in contrast to other CO2 insertion reactions, where CO2–metal interactions have been predicted. Second, the substrates show an unusual coordination mode during CO2 insertion, with the nucleophilic carbon positioned up to 3.6 Å away from rhodium. In order to understand the experimentally observed substrate preferences, we have analyzed a set of five alkenes: an α,β-unsaturated ester, an α,β-unsaturated amide, styrene, and two styrene derivatives. The computational results and additional experiments reported here indicate that the lack of activity with amides is caused by an overly high barrier for CO2 insertion and is not due to catalyst inactivation. Our experimental studies also reveal two putative side reactions, involving oxidative cleavage or dimerization of the alkene substrate. In the presence of CO2, these alternative reaction pathways are suppressed. The overall insights may be relevant for the design of future hydrocarboxylation catalysts.
中文翻译:

铑催化的加氢羧化反应:机理分析揭示了碳-碳键形成的异常过渡态
利用密度泛函理论(PBE-D2 / IEFPCM),研究了铑COD催化苯乙烯衍生物和α,β-不饱和羰基化合物与CO 2的加氢羧化反应的机理。这些计算支持了Mikami及其同事最初提出的催化循环,包括消除β-氢化物,将不饱和底物插入到铑-氢化物键中以及随后用CO 2羧化。发现CO 2插入步骤是速率限制的。计算揭示了两个有趣的方面。首先,在C–CO 2键形成过程中,CO 2分子既不与铑配合物也不与有机锌添加剂相互作用。这似乎与其他CO 2相反插入反应,其中已预测到CO 2与金属的相互作用。其次,在CO 2插入过程中,底物表现出不同寻常的配位模式,亲核碳与铑的位置最高达3.6Å。为了了解实验观察到的底物偏好,我们分析了一组五个烯烃:α,β-不饱和酯,α,β-不饱和酰胺,苯乙烯和两种苯乙烯衍生物。此处报道的计算结果和其他实验表明,酰胺类化合物缺乏活性是由于CO 2的阻隔过高引起的且不是由于催化剂失活引起的。我们的实验研究还揭示了两个假定的副反应,涉及烯烃底物的氧化裂解或二聚化。在CO 2的存在下,这些替代反应途径被抑制。总体见解可能与未来加氢羧化催化剂的设计有关。
更新日期:2018-03-13
中文翻译:
铑催化的加氢羧化反应:机理分析揭示了碳-碳键形成的异常过渡态
利用密度泛函理论(PBE-D2 / IEFPCM),研究了铑COD催化苯乙烯衍生物和α,β-不饱和羰基化合物与CO 2的加氢羧化反应的机理。这些计算支持了Mikami及其同事最初提出的催化循环,包括消除β-氢化物,将不饱和底物插入到铑-氢化物键中以及随后用CO 2羧化。发现CO 2插入步骤是速率限制的。计算揭示了两个有趣的方面。首先,在C–CO 2键形成过程中,CO 2分子既不与铑配合物也不与有机锌添加剂相互作用。这似乎与其他CO 2相反插入反应,其中已预测到CO 2与金属的相互作用。其次,在CO 2插入过程中,底物表现出不同寻常的配位模式,亲核碳与铑的位置最高达3.6Å。为了了解实验观察到的底物偏好,我们分析了一组五个烯烃:α,β-不饱和酯,α,β-不饱和酰胺,苯乙烯和两种苯乙烯衍生物。此处报道的计算结果和其他实验表明,酰胺类化合物缺乏活性是由于CO 2的阻隔过高引起的且不是由于催化剂失活引起的。我们的实验研究还揭示了两个假定的副反应,涉及烯烃底物的氧化裂解或二聚化。在CO 2的存在下,这些替代反应途径被抑制。总体见解可能与未来加氢羧化催化剂的设计有关。


























 京公网安备 11010802027423号
京公网安备 11010802027423号