当前位置:
X-MOL 学术
›
Macromolecules
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Theory and Simulation of Attractive Nanoparticle Transport in Polymer Melts
Macromolecules ( IF 5.5 ) Pub Date : 2018-03-08 00:00:00 , DOI: 10.1021/acs.macromol.7b02694 Umi Yamamoto 1, 2 , Jan-Michael Y. Carrillo , Vera Bocharova , Alexei P. Sokolov 3 , Bobby G. Sumpter , Kenneth S. Schweizer
Macromolecules ( IF 5.5 ) Pub Date : 2018-03-08 00:00:00 , DOI: 10.1021/acs.macromol.7b02694 Umi Yamamoto 1, 2 , Jan-Michael Y. Carrillo , Vera Bocharova , Alexei P. Sokolov 3 , Bobby G. Sumpter , Kenneth S. Schweizer
Affiliation
|
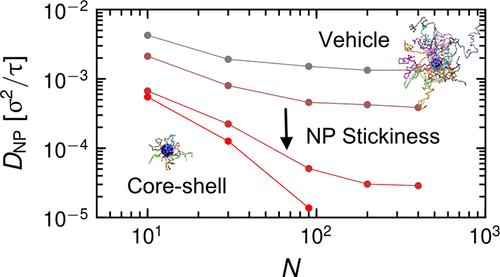
We theoretically study the diffusion of a single attractive nanoparticle (NP) in unentangled and entangled polymer melts based on combining microscopic “core–shell” and “vehicle” mechanisms in a dynamic bond percolation theory framework. A physical picture is constructed which addresses the role of chain length (N), degree of entanglement, nanoparticle size, and NP–polymer attraction strength. The nanoparticle diffusion constant is predicted to initially decrease with N due to the dominance of the core–shell mechanism, then to cross over to the vehicle diffusion regime with a weaker N dependence, and eventually plateau at large enough N. This behavior corresponds to decoupling of NP diffusivity from the macroscopic melt viscosity, which is reminiscent of repulsive NPs in entangled melts, but here it occurs for a distinct physical reason. Specifically, it reflects a crossover to a transport mechanism whereby nanoparticles adsorb on polymer chains and diffuse using them as “vehicles” over a characteristic desorption time scale. Repetition of random desorption events then leads to Fickian long time NP diffusion. Complementary simulations for a range of chain lengths and low to moderate NP–polymer attraction strengths are also performed. They allow testing of the proposed diffusion mechanisms and qualitatively support the theoretically predicted dynamic crossover behavior. When the desorption time is smaller than or comparable to the onset of entangled polymer dynamics, the NP diffusivity becomes almost chain length independent.
中文翻译:

聚合物熔体中有吸引力的纳米颗粒传输的理论和模拟
我们在动态键渗流理论框架内结合微观“核-壳”和“媒介”机理,从理论上研究了单个引诱的纳米粒子(NP)在未缠结和缠结的聚合物熔体中的扩散。构造了一张物理图,处理了链长(N),缠结程度,纳米颗粒尺寸和NP-聚合物吸引力的作用。预计由于核-壳机制的主导,纳米粒子的扩散常数最初会随着N降低,然后以较弱的N依赖性越过载流子扩散机制,最终在足够大的N处达到平稳状态。。此行为对应于NP扩散率与宏观熔体粘度的解耦,这使人联想到缠结熔体中的排斥性NP,但此处发生的原因是明显的物理原因。具体来说,它反映了与传输机制的交叉,纳米颗粒吸附在聚合物链上,并在特定的解吸时间范围内将它们用作“载体”进行扩散。重复随机解吸事件会导致Fickian长时间的NP扩散。还对一系列链长和低至中等的NP-聚合物吸引力进行了补充模拟。它们允许测试所提出的扩散机制,并定性地支持理论上预测的动态交叉行为。当解吸时间小于或等于缠结聚合物动力学的开始时,
更新日期:2018-03-08
中文翻译:
聚合物熔体中有吸引力的纳米颗粒传输的理论和模拟
我们在动态键渗流理论框架内结合微观“核-壳”和“媒介”机理,从理论上研究了单个引诱的纳米粒子(NP)在未缠结和缠结的聚合物熔体中的扩散。构造了一张物理图,处理了链长(N),缠结程度,纳米颗粒尺寸和NP-聚合物吸引力的作用。预计由于核-壳机制的主导,纳米粒子的扩散常数最初会随着N降低,然后以较弱的N依赖性越过载流子扩散机制,最终在足够大的N处达到平稳状态。。此行为对应于NP扩散率与宏观熔体粘度的解耦,这使人联想到缠结熔体中的排斥性NP,但此处发生的原因是明显的物理原因。具体来说,它反映了与传输机制的交叉,纳米颗粒吸附在聚合物链上,并在特定的解吸时间范围内将它们用作“载体”进行扩散。重复随机解吸事件会导致Fickian长时间的NP扩散。还对一系列链长和低至中等的NP-聚合物吸引力进行了补充模拟。它们允许测试所提出的扩散机制,并定性地支持理论上预测的动态交叉行为。当解吸时间小于或等于缠结聚合物动力学的开始时,

























 京公网安备 11010802027423号
京公网安备 11010802027423号