当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Assessment of Real-Time Time-Dependent Density Functional Theory (RT-TDDFT) in Radiation Chemistry: Ionized Water Dimer
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2018-03-07 00:00:00 , DOI: 10.1021/acs.jpca.8b01259 Jan Chalabala 1 , Frank Uhlig 1, 2 , Petr Slavíček 1, 3
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2018-03-07 00:00:00 , DOI: 10.1021/acs.jpca.8b01259 Jan Chalabala 1 , Frank Uhlig 1, 2 , Petr Slavíček 1, 3
Affiliation
|
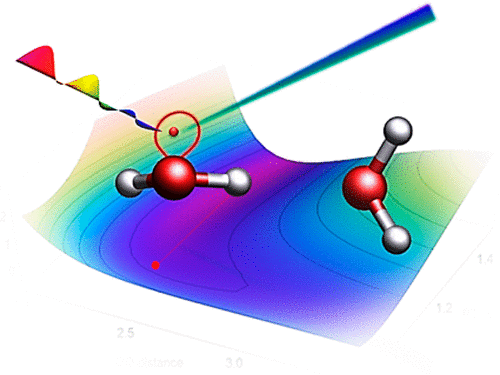
Ionization in the condensed phase and molecular clusters leads to a complicated chain of processes with coupled electron–nuclear dynamics. It is difficult to describe such dynamics with conventional nonadiabatic molecular dynamics schemes since the number of states swiftly increases as the molecular system grows. It is therefore attractive to use a direct electron and nuclear propagation such as the real-time time-dependent density functional theory (RT-TDDFT). Here we report a RT-TDDFT benchmark study on simulations of singly and doubly ionized states of a water monomer and dimer as a prototype for more complex processes in a condensed phase. We employed the RT-TDDFT based Ehrenfest molecular dynamics with a generalized gradient approximate (GGA) functional and compared it with wave-function-based surface hopping (SH) simulations. We found that the initial dynamics of a singly HOMO ionized water dimer is similar for both the RT-TDDFT/GGA and the SH simulations but leads to completely different reaction channels on a longer time scale. This failure is attributed to the self-interaction error in the GGA functionals and it can be avoided by using hybrid functionals with large fraction of exact exchange (represented here by the BHandHLYP functional). The simulations of doubly ionized states are reasonably described already at the GGA level. This suggests that the RT-TDDFT/GGA method could describe processes following the autoionization processes such as Auger emission, while its applicability to more complex processes such as intermolecular Coulombic decay remains limited.
中文翻译:

辐射化学中实时时变密度泛函理论(RT-TDDFT)的评估:电离水二聚体
凝聚相和分子簇中的电离导致电子核动力学耦合的复杂过程链。用常规的非绝热分子动力学方案难以描述这种动力学,因为随着分子系统的增长,状态数迅速增加。因此,使用直接的电子和核传播(例如实时随时间变化的密度泛函理论(RT-TDDFT))是有吸引力的。在这里,我们报告了RT-TDDFT基准研究,该模拟研究了水单体和二聚体的单电离和双电离状态的模拟,作为冷凝阶段中更复杂过程的原型。我们将基于RT-TDDFT的Ehrenfest分子动力学与广义梯度近似(GGA)功能结合起来,并将其与基于波函数的表面跳变(SH)模拟进行了比较。我们发现,对于RT-TDDFT / GGA和SH模拟,单个HOMO离子化水二聚体的初始动力学相似,但在更长的时间范围内导致完全不同的反应通道。该故障归因于GGA功能中的自交互错误,可以通过使用具有大量精确交换(在此以BHandHLYP功能表示)的混合功能来避免。已经在GGA级别上合理地描述了双电离态的模拟。这表明RT-TDDFT / GGA方法可以描述自电离过程(如俄歇发射)之后的过程,而其对更复杂过程(如分子间库仑衰变)的适用性仍然有限。
更新日期:2018-03-07
中文翻译:
辐射化学中实时时变密度泛函理论(RT-TDDFT)的评估:电离水二聚体
凝聚相和分子簇中的电离导致电子核动力学耦合的复杂过程链。用常规的非绝热分子动力学方案难以描述这种动力学,因为随着分子系统的增长,状态数迅速增加。因此,使用直接的电子和核传播(例如实时随时间变化的密度泛函理论(RT-TDDFT))是有吸引力的。在这里,我们报告了RT-TDDFT基准研究,该模拟研究了水单体和二聚体的单电离和双电离状态的模拟,作为冷凝阶段中更复杂过程的原型。我们将基于RT-TDDFT的Ehrenfest分子动力学与广义梯度近似(GGA)功能结合起来,并将其与基于波函数的表面跳变(SH)模拟进行了比较。我们发现,对于RT-TDDFT / GGA和SH模拟,单个HOMO离子化水二聚体的初始动力学相似,但在更长的时间范围内导致完全不同的反应通道。该故障归因于GGA功能中的自交互错误,可以通过使用具有大量精确交换(在此以BHandHLYP功能表示)的混合功能来避免。已经在GGA级别上合理地描述了双电离态的模拟。这表明RT-TDDFT / GGA方法可以描述自电离过程(如俄歇发射)之后的过程,而其对更复杂过程(如分子间库仑衰变)的适用性仍然有限。


























 京公网安备 11010802027423号
京公网安备 11010802027423号