当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Solvent Effects on Electronically Excited States: QM/Continuum Versus QM/Explicit Models
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2018-02-26 00:00:00 , DOI: 10.1021/acs.jpcb.7b12560 Martina De Vetta 1, 2 , Maximilian F. S. J. Menger 1, 3 , Juan J. Nogueira 1 , Leticia González 1
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2018-02-26 00:00:00 , DOI: 10.1021/acs.jpcb.7b12560 Martina De Vetta 1, 2 , Maximilian F. S. J. Menger 1, 3 , Juan J. Nogueira 1 , Leticia González 1
Affiliation
|
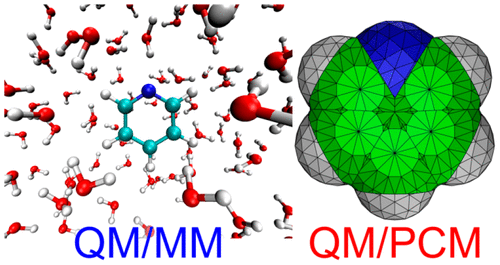
The inclusion of solvent effects in the calculation of excited states is vital to obtain reliable absorption spectra and density of states of solvated chromophores. Here we analyze the performance of three classical approaches to describe aqueous solvent in the calculation of the absorption spectra and density of states of pyridine, tropone, and tropothione. Specifically, we compare the results obtained from quantum mechanics/polarizable continuum model (QM/PCM) versus quantum mechanics/molecular mechanics (QM/MM) in its electrostatic-embedding (QM/MMee) and polarizable-embedding (QM/MMpol) fashions, against full-QM computations, in which the solvent is described at the same level of theory as the chromophore. We show that QM/PCM provides very accurate results describing the excitation energies of ππ* and nπ* transitions, the last ones dominated by strong hydrogen-bonding effects, for the three chromophores. The QM/MMee approach also performs very well for both types of electronic transitions, although the description of the ππ* ones is slightly worse than that obtained from QM/PCM. The QM/MMpol approach performs as well as QM/PCM for describing the energy of ππ* states, but it is not able to provide a satisfactory description of hydrogen-bonding effects on the nπ* states of pyridine and tropone. The relative intensity of the absorption bands is better accounted for by the explicit-solvent models than by the continuum-solvent approach.
中文翻译:

对电子激发态的溶剂影响:QM /连续谱与QM /显式模型
在激发态的计算中包括溶剂效应对于获得可靠的吸收光谱和溶剂化发色团的状态密度至关重要。在这里,我们分析了三种描述水性溶剂的经典方法的性能,这些方法描述了吡啶,托普酮和对硫磷的吸收光谱和状态密度的计算。具体来说,我们比较了从量子力学/极化连续体模型(QM / PCM)与量子力学/分子力学(QM / MM)的静电嵌入(QM / MMee)和极化极化(QM / MMpol)方式获得的结果,而不是全QM计算,其中以与生色团相同的理论水平描述溶剂。我们证明QM / PCM提供了非常准确的结果,描述了ππ*和nπ*跃迁的激发能,对于三个生色团,最后一个主要由强氢键作用主导。尽管对ππ*的描述比从QM / PCM获得的描述稍差,但是QM / MMee方法对两种类型的电子跃迁也都表现良好。QM / MMpol方法的性能与描述ππ*态能量的QM / PCM一样好,但是它不能提供令人满意的氢键对吡啶和对苯二酚nπ*态的氢键效应的描述。显式溶剂模型比连续介质方法更好地解释了吸收带的相对强度。尽管对ππ*的描述比从QM / PCM获得的描述稍差。QM / MMpol方法的性能与描述ππ*态能量的QM / PCM一样好,但是它不能提供令人满意的氢键对吡啶和对苯二酚nπ*态的氢键效应的描述。显式溶剂模型比连续介质方法更好地解释了吸收带的相对强度。尽管对ππ*的描述比从QM / PCM获得的描述稍差。QM / MMpol方法的性能与描述ππ*态能量的QM / PCM一样好,但是它不能提供令人满意的氢键对吡啶和对苯二酚nπ*态的氢键效应的描述。显式溶剂模型比连续介质方法更好地解释了吸收带的相对强度。
更新日期:2018-02-26
中文翻译:
对电子激发态的溶剂影响:QM /连续谱与QM /显式模型
在激发态的计算中包括溶剂效应对于获得可靠的吸收光谱和溶剂化发色团的状态密度至关重要。在这里,我们分析了三种描述水性溶剂的经典方法的性能,这些方法描述了吡啶,托普酮和对硫磷的吸收光谱和状态密度的计算。具体来说,我们比较了从量子力学/极化连续体模型(QM / PCM)与量子力学/分子力学(QM / MM)的静电嵌入(QM / MMee)和极化极化(QM / MMpol)方式获得的结果,而不是全QM计算,其中以与生色团相同的理论水平描述溶剂。我们证明QM / PCM提供了非常准确的结果,描述了ππ*和nπ*跃迁的激发能,对于三个生色团,最后一个主要由强氢键作用主导。尽管对ππ*的描述比从QM / PCM获得的描述稍差,但是QM / MMee方法对两种类型的电子跃迁也都表现良好。QM / MMpol方法的性能与描述ππ*态能量的QM / PCM一样好,但是它不能提供令人满意的氢键对吡啶和对苯二酚nπ*态的氢键效应的描述。显式溶剂模型比连续介质方法更好地解释了吸收带的相对强度。尽管对ππ*的描述比从QM / PCM获得的描述稍差。QM / MMpol方法的性能与描述ππ*态能量的QM / PCM一样好,但是它不能提供令人满意的氢键对吡啶和对苯二酚nπ*态的氢键效应的描述。显式溶剂模型比连续介质方法更好地解释了吸收带的相对强度。尽管对ππ*的描述比从QM / PCM获得的描述稍差。QM / MMpol方法的性能与描述ππ*态能量的QM / PCM一样好,但是它不能提供令人满意的氢键对吡啶和对苯二酚nπ*态的氢键效应的描述。显式溶剂模型比连续介质方法更好地解释了吸收带的相对强度。

























 京公网安备 11010802027423号
京公网安备 11010802027423号