当前位置:
X-MOL 学术
›
Chem. Rev.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Ab Initio Nonadiabatic Quantum Molecular Dynamics
Chemical Reviews ( IF 62.1 ) Pub Date : 2018-02-21 00:00:00 , DOI: 10.1021/acs.chemrev.7b00423 Basile F. E. Curchod 1 , Todd J. Martínez 2, 3
Chemical Reviews ( IF 62.1 ) Pub Date : 2018-02-21 00:00:00 , DOI: 10.1021/acs.chemrev.7b00423 Basile F. E. Curchod 1 , Todd J. Martínez 2, 3
Affiliation
|
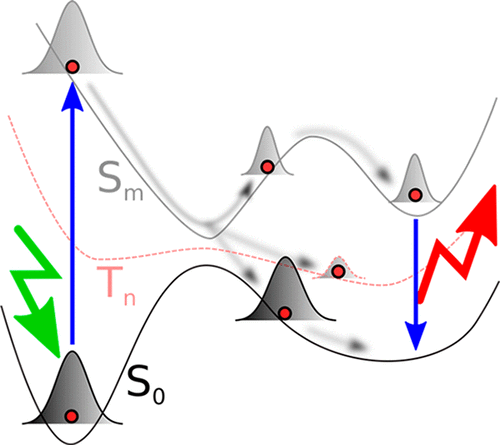
The Born–Oppenheimer approximation underlies much of chemical simulation and provides the framework defining the potential energy surfaces that are used for much of our pictorial understanding of chemical phenomena. However, this approximation breaks down when the dynamics of molecules in excited electronic states are considered. Describing dynamics when the Born–Oppenheimer approximation breaks down requires a quantum mechanical description of the nuclei. Chemical reaction dynamics on excited electronic states is critical for many applications in renewable energy, chemical synthesis, and bioimaging. Furthermore, it is necessary in order to connect with many ultrafast pump–probe spectroscopic experiments. In this review, we provide an overview of methods that can describe nonadiabatic dynamics, with emphasis on those that are able to simultaneously address the quantum mechanics of both electrons and nuclei. Such ab initio quantum molecular dynamics methods solve the electronic Schrödinger equation alongside the nuclear dynamics and thereby avoid the need for precalculation of potential energy surfaces and nonadiabatic coupling matrix elements. Two main families of methods are commonly employed to simulate nonadiabatic dynamics in molecules: full quantum dynamics, such as the multiconfigurational time-dependent Hartree method, and classical trajectory-based approaches, such as trajectory surface hopping. In this review, we describe a third class of methods that is intermediate between the two: Gaussian basis set expansions built around trajectories.
中文翻译:

从头算起非绝热量子分子动力学
Born–Oppenheimer逼近是许多化学模拟的基础,并提供了定义势能面的框架,这些面被用于我们对化学现象的许多图形理解。但是,当考虑激发电子态下的分子动力学时,这种近似分解。描述当Born–Oppenheimer近似分解时的动力学过程需要对原子核进行量子力学描述。激发电子态上的化学反应动力学对于可再生能源,化学合成和生物成像中的许多应用至关重要。此外,有必要连接许多超快泵浦-探针光谱实验。在这篇评论中,我们概述了可以描述非绝热动力学的方法,重点介绍那些能够同时解决电子和原子核的量子力学的技术。这种从头算起的量子分子动力学方法与核动力学一起解决了电子薛定ding方程,从而避免了对势能面和非绝热耦合矩阵元素进行预先计算的需要。通常使用两种主要方法来模拟分子中的非绝热动力学:全量子动力学,例如多配置时间相关的Hartree方法,以及经典的基于轨迹的方法,例如轨迹表面跳变。在本文中,我们描述了介于两者之间的第三类方法:围绕轨迹建立的高斯基集展开。这种从头算起的量子分子动力学方法与核动力学一起解决了电子薛定ding方程,从而避免了对势能面和非绝热耦合矩阵元素进行预先计算的需要。通常使用两种主要方法来模拟分子中的非绝热动力学:全量子动力学,例如多配置时间相关的Hartree方法,以及经典的基于轨迹的方法,例如轨迹表面跳变。在本文中,我们描述了介于两者之间的第三类方法:围绕轨迹建立的高斯基集展开。这种从头算起的量子分子动力学方法与核动力学一起解决了电子薛定ding方程,从而避免了对势能面和非绝热耦合矩阵元素进行预先计算的需要。通常使用两种主要方法来模拟分子中的非绝热动力学:全量子动力学,例如多配置时间相关的Hartree方法,以及经典的基于轨迹的方法,例如轨迹表面跳变。在本文中,我们描述了介于两者之间的第三类方法:围绕轨迹建立的高斯基集展开。
更新日期:2018-02-21
中文翻译:
从头算起非绝热量子分子动力学
Born–Oppenheimer逼近是许多化学模拟的基础,并提供了定义势能面的框架,这些面被用于我们对化学现象的许多图形理解。但是,当考虑激发电子态下的分子动力学时,这种近似分解。描述当Born–Oppenheimer近似分解时的动力学过程需要对原子核进行量子力学描述。激发电子态上的化学反应动力学对于可再生能源,化学合成和生物成像中的许多应用至关重要。此外,有必要连接许多超快泵浦-探针光谱实验。在这篇评论中,我们概述了可以描述非绝热动力学的方法,重点介绍那些能够同时解决电子和原子核的量子力学的技术。这种从头算起的量子分子动力学方法与核动力学一起解决了电子薛定ding方程,从而避免了对势能面和非绝热耦合矩阵元素进行预先计算的需要。通常使用两种主要方法来模拟分子中的非绝热动力学:全量子动力学,例如多配置时间相关的Hartree方法,以及经典的基于轨迹的方法,例如轨迹表面跳变。在本文中,我们描述了介于两者之间的第三类方法:围绕轨迹建立的高斯基集展开。这种从头算起的量子分子动力学方法与核动力学一起解决了电子薛定ding方程,从而避免了对势能面和非绝热耦合矩阵元素进行预先计算的需要。通常使用两种主要方法来模拟分子中的非绝热动力学:全量子动力学,例如多配置时间相关的Hartree方法,以及经典的基于轨迹的方法,例如轨迹表面跳变。在本文中,我们描述了介于两者之间的第三类方法:围绕轨迹建立的高斯基集展开。这种从头算起的量子分子动力学方法与核动力学一起解决了电子薛定ding方程,从而避免了对势能面和非绝热耦合矩阵元素进行预先计算的需要。通常使用两种主要方法来模拟分子中的非绝热动力学:全量子动力学,例如多配置时间相关的Hartree方法,以及经典的基于轨迹的方法,例如轨迹表面跳变。在本文中,我们描述了介于两者之间的第三类方法:围绕轨迹建立的高斯基集展开。


























 京公网安备 11010802027423号
京公网安备 11010802027423号