当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
CAMERRA: An analysis tool for the computation of conformational dynamics by evaluating residue-residue associations
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-02-21 , DOI: 10.1002/jcc.25192 Quentin R. Johnson 1, 2 , Richard J. Lindsay 2, 3 , Tongye Shen 2, 4
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-02-21 , DOI: 10.1002/jcc.25192 Quentin R. Johnson 1, 2 , Richard J. Lindsay 2, 3 , Tongye Shen 2, 4
Affiliation
|
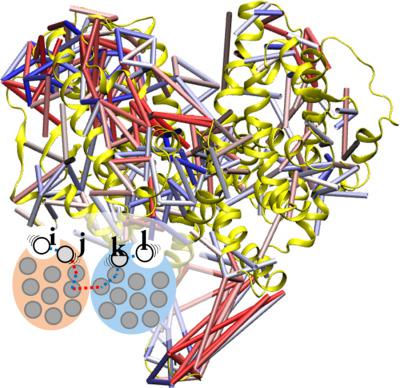
A computational method which extracts the dominant motions from an ensemble of biomolecular conformations via a correlation analysis of residue–residue contacts is presented. The algorithm first renders the structural information into contact matrices, then constructs the collective modes based on the correlated dynamics of a selected set of dynamic contacts. Associated programs can bridge the results for further visualization using graphics software. The aim of this method is to provide an analysis of conformations of biopolymers from the contact viewpoint. It may assist a systematical uncovering of conformational switching mechanisms existing in proteins and biopolymer systems in general by statistical analysis of simulation snapshots. In contrast to conventional correlation analyses of Cartesian coordinates (such as distance covariance analysis and Cartesian principal component analysis), this program also provides an alternative way to locate essential collective motions in general. Herein, we detail the algorithm in a stepwise manner and comment on the importance of the method as applied to decoding allosteric mechanisms. © 2018 Wiley Periodicals, Inc.
中文翻译:

CAMERRA:通过评估残基-残基关联来计算构象动力学的分析工具
提出了一种通过残基-残基接触的相关分析从生物分子构象集合中提取主要运动的计算方法。该算法首先将结构信息呈现为接触矩阵,然后根据选定的一组动态接触的相关动力学构造集体模式。相关程序可以桥接结果,以便使用图形软件进一步可视化。该方法的目的是从接触的角度分析生物聚合物的构象。通过模拟快照的统计分析,它可以帮助系统地揭示蛋白质和生物聚合物系统中存在的构象转换机制。与笛卡尔坐标的传统相关分析(例如距离协方差分析和笛卡尔主成分分析)相比,该程序还提供了一种定位基本集体运动的替代方法。在这里,我们以逐步的方式详细介绍了该算法,并评论了该方法应用于解码变构机制的重要性。© 2018 Wiley Periodicals, Inc.
更新日期:2018-02-21
中文翻译:
CAMERRA:通过评估残基-残基关联来计算构象动力学的分析工具
提出了一种通过残基-残基接触的相关分析从生物分子构象集合中提取主要运动的计算方法。该算法首先将结构信息呈现为接触矩阵,然后根据选定的一组动态接触的相关动力学构造集体模式。相关程序可以桥接结果,以便使用图形软件进一步可视化。该方法的目的是从接触的角度分析生物聚合物的构象。通过模拟快照的统计分析,它可以帮助系统地揭示蛋白质和生物聚合物系统中存在的构象转换机制。与笛卡尔坐标的传统相关分析(例如距离协方差分析和笛卡尔主成分分析)相比,该程序还提供了一种定位基本集体运动的替代方法。在这里,我们以逐步的方式详细介绍了该算法,并评论了该方法应用于解码变构机制的重要性。© 2018 Wiley Periodicals, Inc.

























 京公网安备 11010802027423号
京公网安备 11010802027423号