当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Machine Learning of Partial Charges Derived from High-Quality Quantum-Mechanical Calculations
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-02-20 00:00:00 , DOI: 10.1021/acs.jcim.7b00663 Patrick Bleiziffer 1 , Kay Schaller 1 , Sereina Riniker 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-02-20 00:00:00 , DOI: 10.1021/acs.jcim.7b00663 Patrick Bleiziffer 1 , Kay Schaller 1 , Sereina Riniker 1
Affiliation
|
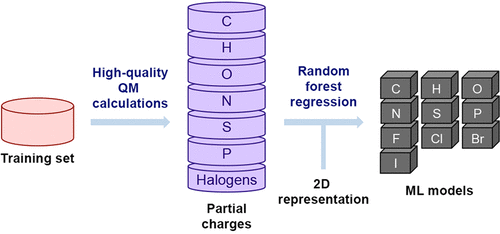
Parametrization of small organic molecules for classical molecular dynamics simulations is not trivial. The vastness of the chemical space makes approaches using building blocks challenging. The most common approach is therefore an individual parametrization of each compound by deriving partial charges from semiempirical or ab initio calculations and inheriting the bonded and van der Waals (Lennard-Jones) parameters from a (bio)molecular force field. The quality of the partial charges generated in this fashion depends on the level of the quantum-chemical calculation as well as on the extraction procedure used. Here, we present a machine learning (ML) based approach for predicting partial charges extracted from density functional theory (DFT) electron densities. The training set was chosen with the goal to provide a broad coverage of the known chemical space of druglike molecules. In addition to the speed of the approach, the partial charges predicted by ML are not dependent on the three-dimensional conformation in contrast to the ones obtained by fitting to the electrostatic potential (ESP). To assess the quality and compatibility with standard force fields, we performed benchmark calculations for the free energy of hydration and liquid properties such as density and heat of vaporization.
中文翻译:

高质量量子力学计算衍生的部分电荷的机器学习
用于经典分子动力学模拟的有机小分子的参数化并非易事。化学空间的广阔性使使用构建模块的方法具有挑战性。因此,最常见的方法是通过从半经验或从头算计算中得出部分电荷,并从(生物)分子力场继承键合和范德华斯(Lennard-Jones)参数来对每种化合物进行单独的参数化。以这种方式产生的部分电荷的质量取决于量子化学计算的水平以及所使用的提取程序。在这里,我们提出了一种基于机器学习(ML)的方法,用于预测从密度泛函理论(DFT)电子密度提取的部分电荷。选择训练集的目的是要广泛涵盖类药物分子的已知化学空间。除了逼近速度之外,与通过拟合静电势(ESP)获得的电荷相反,由ML预测的部分电荷不依赖于三维构象。为了评估质量和与标准力场的兼容性,我们对水合自由能和液体性质(例如密度和汽化热)进行了基准计算。
更新日期:2018-02-20
中文翻译:
高质量量子力学计算衍生的部分电荷的机器学习
用于经典分子动力学模拟的有机小分子的参数化并非易事。化学空间的广阔性使使用构建模块的方法具有挑战性。因此,最常见的方法是通过从半经验或从头算计算中得出部分电荷,并从(生物)分子力场继承键合和范德华斯(Lennard-Jones)参数来对每种化合物进行单独的参数化。以这种方式产生的部分电荷的质量取决于量子化学计算的水平以及所使用的提取程序。在这里,我们提出了一种基于机器学习(ML)的方法,用于预测从密度泛函理论(DFT)电子密度提取的部分电荷。选择训练集的目的是要广泛涵盖类药物分子的已知化学空间。除了逼近速度之外,与通过拟合静电势(ESP)获得的电荷相反,由ML预测的部分电荷不依赖于三维构象。为了评估质量和与标准力场的兼容性,我们对水合自由能和液体性质(例如密度和汽化热)进行了基准计算。


























 京公网安备 11010802027423号
京公网安备 11010802027423号