当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Semiempirical configuration interaction calculations for ru-centered dyes*
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-02-15 , DOI: 10.1002/jcc.25190 Lisa A. Fredin 1 , Thomas C. Allison 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2018-02-15 , DOI: 10.1002/jcc.25190 Lisa A. Fredin 1 , Thomas C. Allison 1
Affiliation
|
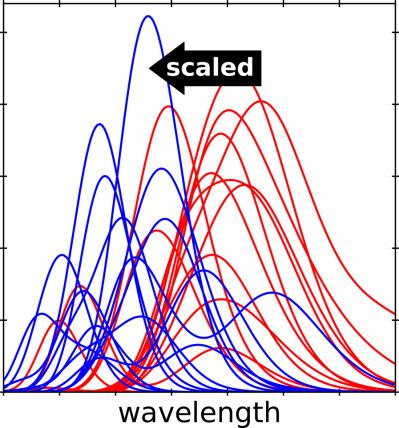
Computational investigation of the photochemical properties of transition‐metal‐centered dyes typically involves optimization of the molecular structure followed by calculation of the UV/visible spectrum. At present, these steps are usually carried out using density functional theory (DFT) and time‐dependent DFT calculations. Recently, we demonstrated that semiempirical methods with appropriate parameterization could yield geometries that were in very good agreement with DFT calculations, allowing large sets of molecules to be screened quickly and efficiently. In this article, we modify a configuration interaction (CI) method based on a semiempirical PM6 Hamiltonian to determine the UV/visible absorption spectra of Ru‐centered complexes. Our modification to the CI method is based on a scaling of the two‐center, two‐electron Coulomb integrals. This modified, PM6‐based method shows a significantly better match to the experimental absorption spectra versus the default configuration interaction method (in MOPAC) on a training set of 13 molecules. In particular, the modified PM6 method blue‐shifts the location of the metal‐to‐ligand charge‐transfer (MLCT) peaks, in better agreement with experimental and DFT‐based computational results, correcting a significant deficiency of the unmodified method. Published 2018. This article is a U.S. Government work and is in the public domain in the USA
中文翻译:

以 ru 为中心的染料的半经验构型相互作用计算*
过渡金属中心染料光化学性质的计算研究通常涉及优化分子结构,然后计算紫外/可见光谱。目前,这些步骤通常使用密度泛函理论(DFT)和时间相关的 DFT 计算来进行。最近,我们证明了具有适当参数化的半经验方法可以产生与 DFT 计算非常一致的几何形状,从而可以快速有效地筛选大量分子。在本文中,我们修改了基于半经验 PM6 哈密顿量的构型相互作用 (CI) 方法,以确定以 Ru 为中心的配合物的紫外/可见吸收光谱。我们对 CI 方法的修改基于双中心、双电子库仑积分的缩放。与默认配置相互作用方法(在 MOPAC 中)相比,这种基于 PM6 的改进方法在 13 个分子的训练集上显示出与实验吸收光谱的明显更好的匹配。特别是,改进后的 PM6 方法使金属-配体电荷转移(MLCT)峰的位置发生蓝移,与实验和基于 DFT 的计算结果更加吻合,纠正了未改进方法的显着缺陷。发表于 2018 年。本文为美国政府作品,在美国属于公有领域 修改后的 PM6 方法使金属到配体电荷转移 (MLCT) 峰的位置发生蓝移,与实验和基于 DFT 的计算结果更加一致,纠正了未修改方法的显着缺陷。发表于 2018 年。本文为美国政府作品,在美国属于公有领域 修改后的 PM6 方法使金属到配体电荷转移 (MLCT) 峰的位置发生蓝移,与实验和基于 DFT 的计算结果更加一致,纠正了未修改方法的显着缺陷。发表于 2018 年。本文为美国政府作品,在美国属于公有领域
更新日期:2018-02-15
中文翻译:
以 ru 为中心的染料的半经验构型相互作用计算*
过渡金属中心染料光化学性质的计算研究通常涉及优化分子结构,然后计算紫外/可见光谱。目前,这些步骤通常使用密度泛函理论(DFT)和时间相关的 DFT 计算来进行。最近,我们证明了具有适当参数化的半经验方法可以产生与 DFT 计算非常一致的几何形状,从而可以快速有效地筛选大量分子。在本文中,我们修改了基于半经验 PM6 哈密顿量的构型相互作用 (CI) 方法,以确定以 Ru 为中心的配合物的紫外/可见吸收光谱。我们对 CI 方法的修改基于双中心、双电子库仑积分的缩放。与默认配置相互作用方法(在 MOPAC 中)相比,这种基于 PM6 的改进方法在 13 个分子的训练集上显示出与实验吸收光谱的明显更好的匹配。特别是,改进后的 PM6 方法使金属-配体电荷转移(MLCT)峰的位置发生蓝移,与实验和基于 DFT 的计算结果更加吻合,纠正了未改进方法的显着缺陷。发表于 2018 年。本文为美国政府作品,在美国属于公有领域 修改后的 PM6 方法使金属到配体电荷转移 (MLCT) 峰的位置发生蓝移,与实验和基于 DFT 的计算结果更加一致,纠正了未修改方法的显着缺陷。发表于 2018 年。本文为美国政府作品,在美国属于公有领域 修改后的 PM6 方法使金属到配体电荷转移 (MLCT) 峰的位置发生蓝移,与实验和基于 DFT 的计算结果更加一致,纠正了未修改方法的显着缺陷。发表于 2018 年。本文为美国政府作品,在美国属于公有领域


























 京公网安备 11010802027423号
京公网安备 11010802027423号