当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Statistical Model To Decipher Protein Folding/Unfolding at a Local Scale
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2018-02-15 00:00:00 , DOI: 10.1021/acs.jpcb.7b10733 Paul Grassein 1 , Patrice Delarue 1 , Harold A. Scheraga 2 , Gia G. Maisuradze 2 , Patrick Senet 1, 2
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2018-02-15 00:00:00 , DOI: 10.1021/acs.jpcb.7b10733 Paul Grassein 1 , Patrice Delarue 1 , Harold A. Scheraga 2 , Gia G. Maisuradze 2 , Patrick Senet 1, 2
Affiliation
|
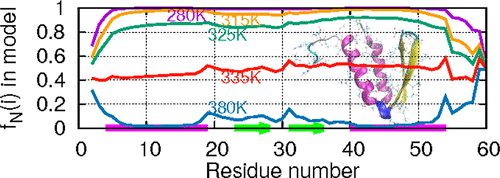
Protein folding/unfolding can be analyzed experimentally at a local scale by monitoring the physical properties of local probes as a function of the temperature, for example, the distance between fluorophores or the values of chemical shifts of backbone atoms. Here, the analytical Lifson–Roig model for the helix–coil transition is modified to analyze local thermal unfolding of the fast-folder W protein of bacteriophage lambda (gpW) simulated by all-atom molecular dynamics (MD) simulations in explicit solvent at 15 different temperatures. The protein structure is described by the coarse-grained dihedral angles (γ) and bond angles (θ) built between successive Cα–Cα virtual bonds. Each (γ,θ) pair serves as a local probe of protein unfolding. Local native/non-native states are defined for each pair of (γ,θ) angles by analyzing the free-energy landscapes ΔG(γ,θ) computed from MD trajectories. The three local elementary equilibrium constants of the model are extracted for each (γ,θ) pair along the sequence from MD simulations, and the model predictions are compared to the MD data. Using only the local equilibrium constants as an input, we show that the local denaturation curves computed from the model partition function fit their MD simulated counterparts in a satisfying manner without any adjustment. In the model and MD simulations, gpW unfolds gradually between 320 and 340 K, with an average native percentage decreasing from 0.8 (320 K) to 0.2 (340 K). In the prism of the model, there is no stable structure at the local scale in this 20 K unfolding temperature range. The enthalpy change upon local unfolding computed from the model and from MD trajectories suggests that the unfolded state between 320 and 340 K corresponds to a dynamical equilibrium between a large ensemble of constantly changing structures. The present results confirm the downhill unfolding of gpW, which does not obey a two-state global folding/unfolding model, and shed light on the interpretation of local denaturation curves.
中文翻译:

统计模型,以解密蛋白质在本地规模的折叠/展开。
通过监测作为温度的函数的局部探针的物理特性(例如,荧光团之间的距离或骨架原子的化学位移值),可以在局部规模上通过实验分析蛋白质折叠/展开。在这里,修改了螺旋-螺旋转变的解析Lifson-Roig模型,以分析全噬菌体分子动力学(MD)在显式溶剂中于15时模拟的噬菌体λ(gpW)的快速文件夹W蛋白的局部热解折叠。不同的温度。该蛋白质结构由连续℃之间建立的粗粒二面角(γ)和键角(θ)描述α -C α虚拟债券。每个(γ,θ)对都充当蛋白质展开的局部探针。通过分析自由能态势ΔG为每对(γ,θ)角定义局部自然/非自然状态从MD轨迹计算出的(γ,θ)。沿着MD仿真中的序列为每个(γ,θ)对提取模型的三个局部基本平衡常数,并将模型预测与MD数据进行比较。仅使用局部平衡常数作为输入,我们表明从模型分区函数计算出的局部变性曲线以令人满意的方式拟合了其MD模拟对应物,而无需进行任何调整。在模型和MD仿真中,gpW在320和340 K之间逐渐展开,平均原始百分比从0.8(320 K)降低到0.2(340 K)。在该模型的棱镜中,在此20 K展开温度范围内,在局部尺度上没有稳定的结构。根据模型和MD轨迹计算出的局部展开时的焓变表明,在320 K和340 K之间的展开状态对应于不断变化的结构大集合之间的动力学平衡。目前的结果证实了gpW的下坡展开,它不遵循两态全局折叠/展开模型,并为解释局部变性曲线提供了启示。
更新日期:2018-02-15
中文翻译:
统计模型,以解密蛋白质在本地规模的折叠/展开。
通过监测作为温度的函数的局部探针的物理特性(例如,荧光团之间的距离或骨架原子的化学位移值),可以在局部规模上通过实验分析蛋白质折叠/展开。在这里,修改了螺旋-螺旋转变的解析Lifson-Roig模型,以分析全噬菌体分子动力学(MD)在显式溶剂中于15时模拟的噬菌体λ(gpW)的快速文件夹W蛋白的局部热解折叠。不同的温度。该蛋白质结构由连续℃之间建立的粗粒二面角(γ)和键角(θ)描述α -C α虚拟债券。每个(γ,θ)对都充当蛋白质展开的局部探针。通过分析自由能态势ΔG为每对(γ,θ)角定义局部自然/非自然状态从MD轨迹计算出的(γ,θ)。沿着MD仿真中的序列为每个(γ,θ)对提取模型的三个局部基本平衡常数,并将模型预测与MD数据进行比较。仅使用局部平衡常数作为输入,我们表明从模型分区函数计算出的局部变性曲线以令人满意的方式拟合了其MD模拟对应物,而无需进行任何调整。在模型和MD仿真中,gpW在320和340 K之间逐渐展开,平均原始百分比从0.8(320 K)降低到0.2(340 K)。在该模型的棱镜中,在此20 K展开温度范围内,在局部尺度上没有稳定的结构。根据模型和MD轨迹计算出的局部展开时的焓变表明,在320 K和340 K之间的展开状态对应于不断变化的结构大集合之间的动力学平衡。目前的结果证实了gpW的下坡展开,它不遵循两态全局折叠/展开模型,并为解释局部变性曲线提供了启示。

























 京公网安备 11010802027423号
京公网安备 11010802027423号