当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Metagenomic Taxonomy-Guided Database-Searching Strategy for Improving Metaproteomic Analysis
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2018-02-26 00:00:00 , DOI: 10.1021/acs.jproteome.7b00894 Jinqiu Xiao 1 , Alessandro Tanca 2 , Ben Jia 1 , Runqing Yang 3 , Bo Wang 1 , Yu Zhang 4 , Jing Li 1
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2018-02-26 00:00:00 , DOI: 10.1021/acs.jproteome.7b00894 Jinqiu Xiao 1 , Alessandro Tanca 2 , Ben Jia 1 , Runqing Yang 3 , Bo Wang 1 , Yu Zhang 4 , Jing Li 1
Affiliation
|
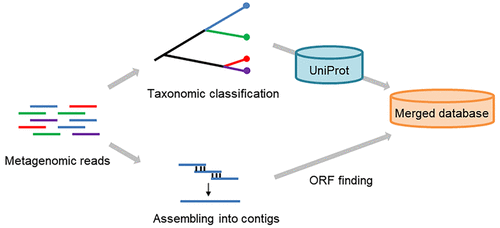
Metaproteomics provides a direct measure of the functional information by investigating all proteins expressed by a microbiota. However, due to the complexity and heterogeneity of microbial communities, it is very hard to construct a sequence database suitable for a metaproteomic study. Using a public database, researchers might not be able to identify proteins from poorly characterized microbial species, while a sequencing-based metagenomic database may not provide adequate coverage for all potentially expressed protein sequences. To address this challenge, we propose a metagenomic taxonomy-guided database-search strategy (MT), in which a merged database is employed, consisting of both taxonomy-guided reference protein sequences from public databases and proteins from metagenome assembly. By applying our MT strategy to a mock microbial mixture, about two times as many peptides were detected as with the metagenomic database only. According to the evaluation of the reliability of taxonomic attribution, the rate of misassignments was comparable to that obtained using an a priori matched database. We also evaluated the MT strategy with a human gut microbial sample, and we found 1.7 times as many peptides as using a standard metagenomic database. In conclusion, our MT strategy allows the construction of databases able to provide high sensitivity and precision in peptide identification in metaproteomic studies, enabling the detection of proteins from poorly characterized species within the microbiota.
中文翻译:

元基因组分类学指导的数据库搜索策略,用于改进蛋白质组学分析
通过研究微生物群表达的所有蛋白质,元蛋白质组学可直接测量功能信息。然而,由于微生物群落的复杂性和异质性,很难构建适合于元蛋白质组学研究的序列数据库。使用公共数据库,研究人员可能无法从特征较差的微生物物种中鉴定蛋白质,而基于测序的宏基因组数据库可能无法为所有潜在表达的蛋白质序列提供足够的覆盖率。为了应对这一挑战,我们提出了一种宏基因组分类学指导的数据库搜索策略(MT),其中采用了合并数据库,该数据库由公共数据库的分类学指导的参考蛋白序列和元基因组组装的蛋白组成。通过将我们的MT策略应用于模拟微生物混合物,与仅使用宏基因组数据库检测到的肽相比,检测到的多肽大约多出两倍。根据对分类学归因的可靠性的评估,误分配率与使用先验匹配数据库获得的误分配率相当。我们还用人类肠道微生物样品评估了MT策略,发现的肽量是使用标准宏基因组学数据库的1.7倍。总之,我们的MT策略允许构建能够在元蛋白质组学研究中的肽段鉴定中提供高灵敏度和精确度的数据库,从而能够从微生物群中特征较弱的物种中检测蛋白质。错误分配的比率与使用先验匹配数据库获得的比率相当。我们还用人类肠道微生物样品评估了MT策略,发现的肽量是使用标准宏基因组学数据库的1.7倍。总之,我们的MT策略允许构建能够在元蛋白质组学研究中的肽段鉴定中提供高灵敏度和精确度的数据库,从而能够从微生物群中特征较弱的物种中检测蛋白质。错误分配的比率与使用先验匹配数据库获得的比率相当。我们还用人类肠道微生物样品评估了MT策略,发现的肽量是使用标准宏基因组学数据库的1.7倍。总之,我们的MT策略允许构建能够在元蛋白质组学研究中的肽段鉴定中提供高灵敏度和精确度的数据库,从而能够从微生物群中特征较弱的物种中检测蛋白质。
更新日期:2018-02-27
中文翻译:
元基因组分类学指导的数据库搜索策略,用于改进蛋白质组学分析
通过研究微生物群表达的所有蛋白质,元蛋白质组学可直接测量功能信息。然而,由于微生物群落的复杂性和异质性,很难构建适合于元蛋白质组学研究的序列数据库。使用公共数据库,研究人员可能无法从特征较差的微生物物种中鉴定蛋白质,而基于测序的宏基因组数据库可能无法为所有潜在表达的蛋白质序列提供足够的覆盖率。为了应对这一挑战,我们提出了一种宏基因组分类学指导的数据库搜索策略(MT),其中采用了合并数据库,该数据库由公共数据库的分类学指导的参考蛋白序列和元基因组组装的蛋白组成。通过将我们的MT策略应用于模拟微生物混合物,与仅使用宏基因组数据库检测到的肽相比,检测到的多肽大约多出两倍。根据对分类学归因的可靠性的评估,误分配率与使用先验匹配数据库获得的误分配率相当。我们还用人类肠道微生物样品评估了MT策略,发现的肽量是使用标准宏基因组学数据库的1.7倍。总之,我们的MT策略允许构建能够在元蛋白质组学研究中的肽段鉴定中提供高灵敏度和精确度的数据库,从而能够从微生物群中特征较弱的物种中检测蛋白质。错误分配的比率与使用先验匹配数据库获得的比率相当。我们还用人类肠道微生物样品评估了MT策略,发现的肽量是使用标准宏基因组学数据库的1.7倍。总之,我们的MT策略允许构建能够在元蛋白质组学研究中的肽段鉴定中提供高灵敏度和精确度的数据库,从而能够从微生物群中特征较弱的物种中检测蛋白质。错误分配的比率与使用先验匹配数据库获得的比率相当。我们还用人类肠道微生物样品评估了MT策略,发现的肽量是使用标准宏基因组学数据库的1.7倍。总之,我们的MT策略允许构建能够在元蛋白质组学研究中的肽段鉴定中提供高灵敏度和精确度的数据库,从而能够从微生物群中特征较弱的物种中检测蛋白质。


























 京公网安备 11010802027423号
京公网安备 11010802027423号