当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Benchmarking lithium amide versus amine bonding by charge density and energy decomposition analysis arguments†
Chemical Science ( IF 8.4 ) Pub Date : 2018-02-08 00:00:00 , DOI: 10.1039/c7sc05368a Felix Engelhardt 1, 2, 3, 4 , Christian Maaß 1, 2, 3, 4 , Diego M. Andrada 4, 5, 6, 7 , Regine Herbst-Irmer 1, 2, 3, 4 , Dietmar Stalke 1, 2, 3, 4
Chemical Science ( IF 8.4 ) Pub Date : 2018-02-08 00:00:00 , DOI: 10.1039/c7sc05368a Felix Engelhardt 1, 2, 3, 4 , Christian Maaß 1, 2, 3, 4 , Diego M. Andrada 4, 5, 6, 7 , Regine Herbst-Irmer 1, 2, 3, 4 , Dietmar Stalke 1, 2, 3, 4
Affiliation
|
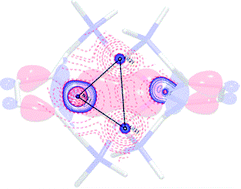
Lithium amides are versatile C–H metallation reagents with vast industrial demand because of their high basicity combined with their weak nucleophilicity, and they are applied in kilotons worldwide annually. The nuclearity of lithium amides, however, modifies and steers reactivity, region- and stereo-selectivity and product diversification in organic syntheses. In this regard, it is vital to understand Li–N bonding as it causes the aggregation of lithium amides to form cubes or ladders from the polar Li–N covalent metal amide bond along the ring stacking and laddering principle. Deaggregation, however, is more governed by the Li←N donor bond to form amine adducts. The geometry of the solid state structures already suggests that there is σ- and π-contribution to the covalent bond. To quantify the mutual influence, we investigated [{(Me2NCH2)2(C4H2N)}Li]2 (1) by means of experimental charge density calculations based on the quantum theory of atoms in molecules (QTAIM) and DFT calculations using energy decomposition analysis (EDA). This new approach allows for the grading of electrostatic Li+N−, covalent Li–N and donating Li←N bonding, and provides a way to modify traditional widely-used heuristic concepts such as the −I and +I inductive effects. The electron density ρ(r) and its second derivative, the Laplacian ∇2ρ(r), mirror the various types of bonding. Most remarkably, from the topological descriptors, there is no clear separation of the lithium amide bonds from the lithium amine donor bonds. The computed natural partial charges for lithium are only +0.58, indicating an optimal density supply from the four nitrogen atoms, while the Wiberg bond orders of about 0.14 au suggest very weak bonding. The interaction energy between the two pincer molecules, (C4H2N)22−, with the Li22+ moiety is very strong (ca. −628 kcal mol−1), followed by the bond dissociation energy (−420.9 kcal mol−1). Partitioning the interaction energy into the Pauli (ΔEPauli), dispersion (ΔEdisp), electrostatic (ΔEelstat) and orbital (ΔEorb) terms gives a 71–72% ionic and 25–26% covalent character of the Li–N bond, different to the old dichotomy of 95 to 5%. In this regard, there is much more potential to steer the reactivity with various substituents and donor solvents than has been anticipated so far.
中文翻译:

通过电荷密度和能量分解分析论据对 酰胺化锂与胺键进行基准测试†
氨基化锂因其高碱性和弱亲核性而成为具有广泛工业需求的通用CH金属化试剂,并且每年在全球范围内以千吨计应用。但是,氨基酰胺锂的核性质会改变并控制有机合成中的反应性,区域选择性和立体选择性以及产物多样化。在这方面,了解Li-N键至关重要,因为它会导致锂酰胺的聚集沿着环堆积和阶梯原理从极性的Li-N共价金属酰胺键形成立方或梯形。然而,解聚更多地受Li←N供体键形成胺加合物的支配。固态结构的几何形状已经表明对共价键有σ和π贡献。为了量化相互影响,我们调查了[{(2 NCH 2) 2(C 4 H 2 N)} Li] 2( 1)通过基于分子中原子的量子理论的实验电荷密度计算(QTAIM)和使用能量分解分析(EDA)的DFT计算来实现。这种新方法允许对静电栗分级+ Ñ - ,共价立-N和供李←N键,和提供了一种方法来修改的传统广泛使用的试探法的概念,如-I和+ I电感效应。电子密度ρ( [R )和它的二阶导数,拉普拉斯∇ 2 ρ( ř),镜像各种类型的粘合。最显着的是,从拓扑描述子来看,没有将酰胺酰胺锂与胺胺供体键清楚地分开。锂的计算的自然部分电荷仅为+0.58,表明来自四个氮原子的最佳密度供给,而约0.14 au的Wiberg键阶表明键非常弱。两个钳分子(C 4 H 2 N)2 2-与Li 2 2+之间的相互作用能非常强(约-628 kcal mol -1),其次是键解离能(-420.9) kcal mol -1)。将相互作用能分配到Pauli(ΔE Pauli),色散(ΔE disp),静电(ΔE elstat)和轨道(ΔE orb)项给出了Li–N键的71–72%离子性和25–26%共价性,这与旧的不同二分法为95%到5%。在这方面,与迄今为止所预期的相比,具有更多的潜力来控制与各种取代基和供体溶剂的反应性。
更新日期:2018-02-08
中文翻译:
通过电荷密度和能量分解分析论据对 酰胺化锂与胺键进行基准测试†
氨基化锂因其高碱性和弱亲核性而成为具有广泛工业需求的通用CH金属化试剂,并且每年在全球范围内以千吨计应用。但是,氨基酰胺锂的核性质会改变并控制有机合成中的反应性,区域选择性和立体选择性以及产物多样化。在这方面,了解Li-N键至关重要,因为它会导致锂酰胺的聚集沿着环堆积和阶梯原理从极性的Li-N共价金属酰胺键形成立方或梯形。然而,解聚更多地受Li←N供体键形成胺加合物的支配。固态结构的几何形状已经表明对共价键有σ和π贡献。为了量化相互影响,我们调查了[{(2 NCH 2) 2(C 4 H 2 N)} Li] 2( 1)通过基于分子中原子的量子理论的实验电荷密度计算(QTAIM)和使用能量分解分析(EDA)的DFT计算来实现。这种新方法允许对静电栗分级+ Ñ - ,共价立-N和供李←N键,和提供了一种方法来修改的传统广泛使用的试探法的概念,如-I和+ I电感效应。电子密度ρ( [R )和它的二阶导数,拉普拉斯∇ 2 ρ( ř),镜像各种类型的粘合。最显着的是,从拓扑描述子来看,没有将酰胺酰胺锂与胺胺供体键清楚地分开。锂的计算的自然部分电荷仅为+0.58,表明来自四个氮原子的最佳密度供给,而约0.14 au的Wiberg键阶表明键非常弱。两个钳分子(C 4 H 2 N)2 2-与Li 2 2+之间的相互作用能非常强(约-628 kcal mol -1),其次是键解离能(-420.9) kcal mol -1)。将相互作用能分配到Pauli(ΔE Pauli),色散(ΔE disp),静电(ΔE elstat)和轨道(ΔE orb)项给出了Li–N键的71–72%离子性和25–26%共价性,这与旧的不同二分法为95%到5%。在这方面,与迄今为止所预期的相比,具有更多的潜力来控制与各种取代基和供体溶剂的反应性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号