Chemical Physics Letters ( IF 2.8 ) Pub Date : 2018-02-03 , DOI: 10.1016/j.cplett.2018.02.005 P. Jasik , T. Kilich , J. Kozicki , J.E. Sienkiewicz
|
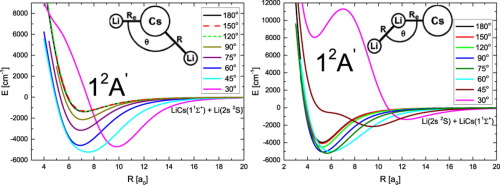
Ab initio quantum chemistry calculations are performed for the mixed alkali triatomic system. Global minima of the ground and first excited doublet states of the trimer are found and Born-Oppenheimer potential energy surfaces of the Li atom interacting with the LiCs molecule were calculated for these states. The lithium atom is placed at various distances and bond angles from the lithium-caesium dimer. Three-body nonadditive forces of the Li2Cs molecule in the global minimum are investigated. Dimer-atom interactions are found to be strongly attractive and may be important in the experiments, particularly involving cold alkali polar dimers.
中文翻译:
Li + LiCs系统低电子态的势能面
从头开始进行量子化学计算,以计算混合碱三原子体系。发现了三聚体的基态和第一激发双峰态的全局极小值,并针对这些态计算了与LiCs分子相互作用的Li原子的Born-Oppenheimer势能面。锂原子与锂铯二聚体的距离和键角不同。研究了Li 2 Cs分子在整体最小值中的三体非相加力。发现二聚体-原子相互作用非常有吸引力,并且在实验中可能很重要,特别是涉及冷碱极性二聚体时。


























 京公网安备 11010802027423号
京公网安备 11010802027423号