当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Weighted Averaging Scheme and Local Atomic Descriptor for pKa Prediction Based on Density Functional Theory
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-02-05 00:00:00 , DOI: 10.1021/acs.jcim.7b00537 Haoyu S. Yu 1 , Mark A. Watson 1 , Art D. Bochevarov 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-02-05 00:00:00 , DOI: 10.1021/acs.jcim.7b00537 Haoyu S. Yu 1 , Mark A. Watson 1 , Art D. Bochevarov 1
Affiliation
|
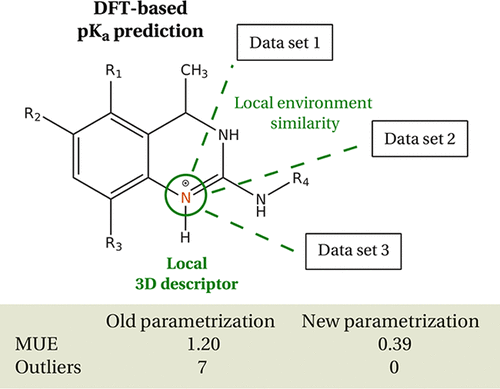
As a continuation of our work on developing a density functional theory-based pKa predictor, we present conceptual improvements to our previously published shell model, which is a hierarchical organization of pKa training sets and which, in principle, covers all chemical space. The improvements concern the way the studied chemical compound is associated with the data points from the training sets. By introducing a new descriptor of the local atomic environment which foregoes dependence on chemical bonding and connectivity, we are able to automatically locate molecules from the training set that are most relevant to the proton dissociation equilibrium under study. This new scheme leads to the prediction of a single pKa value weighted across multiple training sets and thus patches a defect disclosed in the formulation of our previous model. Using the new parametrization approach, the pKa prediction gets rid of outliers reported in previous applications of our approach, eliminates ambiguity in interpreting the results, and improves the overall accuracy. Our new treatment accounts for multiple conformations both on the level of energetics and parametrization. Illustrative results are shown for several types of chemical structures containing guanidine, amidine, amine, and phenol functional groups, and which are representative of practically important large and flexible drug-like molecules. Our method’s performance is compared to the performance of other previously published pKa prediction methods. Further possible improvements to the organization of the training sets and the potential application of our new local atomic descriptor to other kinds of parametrizations are discussed.
中文翻译:

基于密度泛函理论的p K a预测的加权平均方案和局部原子描述符
作为开发基于密度泛函理论的预测因子p K的工作的继续,我们对先前发布的壳模型进行了概念上的改进,壳模型是p K的分层组织,是一个训练集,并且在原则上涵盖所有化学空间。改进涉及将研究的化合物与训练集中的数据点相关联的方式。通过引入一个新的局部原子环境描述子,该描述子不再依赖化学键和连通性,我们能够从训练集中自动定位与所研究的质子解离平衡最相关的分子。这种新方案导致对单个p K a的预测值在多个训练集上加权,从而修补了我们先前模型的公式中披露的缺陷。使用新的参数化方法,p K a预测摆脱了我们方法先前应用中报告的异常值,消除了解释结果的歧义,并提高了总体准确性。我们的新疗法在能量学和参数化方面都考虑了多种构象。显示了几种含有胍、,、胺和苯酚官能团的化学结构的说明性结果,这些化学结构代表了实际上重要的大而柔软的类药物分子。我们的方法的性能与其他先前发布的p K a的性能进行了比较预测方法。讨论了对训练集的组织的进一步可能的改进以及将我们的新局部原子描述符应用于其他类型的参数化的潜在应用。
更新日期:2018-02-05
中文翻译:
基于密度泛函理论的p K a预测的加权平均方案和局部原子描述符
作为开发基于密度泛函理论的预测因子p K的工作的继续,我们对先前发布的壳模型进行了概念上的改进,壳模型是p K的分层组织,是一个训练集,并且在原则上涵盖所有化学空间。改进涉及将研究的化合物与训练集中的数据点相关联的方式。通过引入一个新的局部原子环境描述子,该描述子不再依赖化学键和连通性,我们能够从训练集中自动定位与所研究的质子解离平衡最相关的分子。这种新方案导致对单个p K a的预测值在多个训练集上加权,从而修补了我们先前模型的公式中披露的缺陷。使用新的参数化方法,p K a预测摆脱了我们方法先前应用中报告的异常值,消除了解释结果的歧义,并提高了总体准确性。我们的新疗法在能量学和参数化方面都考虑了多种构象。显示了几种含有胍、,、胺和苯酚官能团的化学结构的说明性结果,这些化学结构代表了实际上重要的大而柔软的类药物分子。我们的方法的性能与其他先前发布的p K a的性能进行了比较预测方法。讨论了对训练集的组织的进一步可能的改进以及将我们的新局部原子描述符应用于其他类型的参数化的潜在应用。


























 京公网安备 11010802027423号
京公网安备 11010802027423号