当前位置:
X-MOL 学术
›
WIREs Comput. Mol. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Periodic and fragment models based on the local correlation approach
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 11.4 ) Pub Date : 2018-01-17 , DOI: 10.1002/wcms.1357 Denis Usvyat 1 , Lorenzo Maschio 2 , Martin Schütz 1
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 11.4 ) Pub Date : 2018-01-17 , DOI: 10.1002/wcms.1357 Denis Usvyat 1 , Lorenzo Maschio 2 , Martin Schütz 1
Affiliation
|
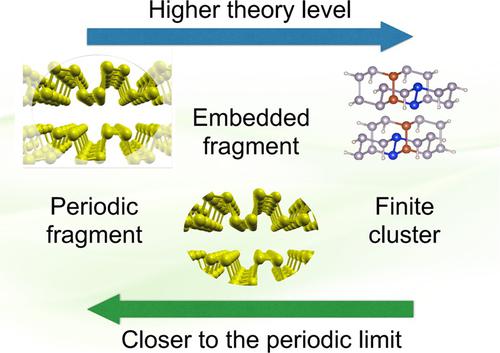
A rigorous treatment of dynamical electron correlation in crystalline solids is one of the main challenges in today's materials quantum chemistry and theoretical solid state physics. In this study, we address this problem by using the local correlation approach and exploring a variety of methods, ranging from the full periodic treatment through embedded fragments to finite clusters. Apart from the computational advantages, the direct‐space local representation for the occupied space allows one to partition the system into fragments and thus forms a natural basis for a hierarchy of embedding models. Furthermore, a subset of localized orbitals in a cluster or a fragment can be chosen to mimic the unit cell of the reference periodic system. Introduction of such subsets allows one to define a formal quantity “the correlation energy per unit cell”, which is directly related to the correlation energy per unit cell in the crystal. The orbital pairs, where neither of the two localized orbital indices belongs to the “unit cell” do not explicitly contribute to the “energy per cell”: Their role is to provide correlated embedding via the couplings in the amplitude equations. The periodic, fragment and finite‐cluster approaches can be combined in a form of high precision computational protocols, where progressively higher‐level corrections are evaluated using lower‐level embedding models. We apply these techniques to investigate the importance of Coulomb screening in dispersively interacting systems on the examples of the phosphorene bilayer and the adsorption of water on 2D silica.
中文翻译:

基于局部相关方法的周期模型和碎片模型
严格处理结晶固体中的动态电子相关性是当今材料量子化学和理论固体物理学的主要挑战之一。在这项研究中,我们通过使用局部相关方法并探索各种方法来解决此问题,从完整的定期处理到嵌入的碎片再到有限的簇。除了计算优势外,所占空间的直接空间局部表示允许将系统划分为多个片段,从而为嵌入模型的层次结构提供了自然的基础。此外,可以选择簇或片段中局部轨道的子集来模拟参考周期系统的晶胞。引入这样的子集可以定义一个正式的量“每单位单元的相关能量”,这直接关系到晶体中每晶胞的相关能量。两个局部轨道索引都不属于“单位单元”的轨道对并没有明确地贡献“每个单元的能量”:它们的作用是通过振幅方程中的耦合提供相关的嵌入。可以采用高精度计算协议的形式来组合周期,片段和有限簇方法,其中使用较低层级的嵌入模型来评估逐步进行的较高层级校正。我们应用这些技术来研究库仑筛查在磷光双层的例子和2D二氧化硅上水的吸附作用的分散相互作用系统中的重要性。其中两个局部轨道索引都不属于“单位晶胞”的情况下,对“每个晶胞的能量”没有明显贡献:它们的作用是通过振幅方程中的耦合提供相关的嵌入。可以采用高精度计算协议的形式来组合周期,片段和有限簇方法,其中使用较低层级的嵌入模型来评估逐步进行的较高层级校正。我们应用这些技术来研究库仑筛查在磷光双层的例子和2D二氧化硅上水的吸附作用的分散相互作用系统中的重要性。其中两个局部轨道索引都不属于“单位晶胞”的情况下,对“每个晶胞的能量”没有明显贡献:它们的作用是通过振幅方程中的耦合提供相关的嵌入。可以采用高精度计算协议的形式来组合周期,片段和有限簇方法,其中使用较低层级的嵌入模型来评估逐步进行的较高层级校正。我们应用这些技术来研究库仑筛查在磷光双层的例子和2D二氧化硅上水的吸附作用的分散相互作用系统中的重要性。片段和有限簇方法可以以高精度计算协议的形式结合使用,其中逐步使用较低级别的嵌入模型来评估较高级别的校正。我们应用这些技术来研究库仑筛查在磷光双层的例子和2D二氧化硅上水的吸附作用的分散相互作用系统中的重要性。片段和有限簇方法可以以高精度计算协议的形式结合使用,其中逐步使用较低级别的嵌入模型来评估较高级别的校正。我们应用这些技术来研究库仑筛查在磷光双层的例子和2D二氧化硅上水的吸附作用的分散相互作用系统中的重要性。
更新日期:2018-01-17
中文翻译:
基于局部相关方法的周期模型和碎片模型
严格处理结晶固体中的动态电子相关性是当今材料量子化学和理论固体物理学的主要挑战之一。在这项研究中,我们通过使用局部相关方法并探索各种方法来解决此问题,从完整的定期处理到嵌入的碎片再到有限的簇。除了计算优势外,所占空间的直接空间局部表示允许将系统划分为多个片段,从而为嵌入模型的层次结构提供了自然的基础。此外,可以选择簇或片段中局部轨道的子集来模拟参考周期系统的晶胞。引入这样的子集可以定义一个正式的量“每单位单元的相关能量”,这直接关系到晶体中每晶胞的相关能量。两个局部轨道索引都不属于“单位单元”的轨道对并没有明确地贡献“每个单元的能量”:它们的作用是通过振幅方程中的耦合提供相关的嵌入。可以采用高精度计算协议的形式来组合周期,片段和有限簇方法,其中使用较低层级的嵌入模型来评估逐步进行的较高层级校正。我们应用这些技术来研究库仑筛查在磷光双层的例子和2D二氧化硅上水的吸附作用的分散相互作用系统中的重要性。其中两个局部轨道索引都不属于“单位晶胞”的情况下,对“每个晶胞的能量”没有明显贡献:它们的作用是通过振幅方程中的耦合提供相关的嵌入。可以采用高精度计算协议的形式来组合周期,片段和有限簇方法,其中使用较低层级的嵌入模型来评估逐步进行的较高层级校正。我们应用这些技术来研究库仑筛查在磷光双层的例子和2D二氧化硅上水的吸附作用的分散相互作用系统中的重要性。其中两个局部轨道索引都不属于“单位晶胞”的情况下,对“每个晶胞的能量”没有明显贡献:它们的作用是通过振幅方程中的耦合提供相关的嵌入。可以采用高精度计算协议的形式来组合周期,片段和有限簇方法,其中使用较低层级的嵌入模型来评估逐步进行的较高层级校正。我们应用这些技术来研究库仑筛查在磷光双层的例子和2D二氧化硅上水的吸附作用的分散相互作用系统中的重要性。片段和有限簇方法可以以高精度计算协议的形式结合使用,其中逐步使用较低级别的嵌入模型来评估较高级别的校正。我们应用这些技术来研究库仑筛查在磷光双层的例子和2D二氧化硅上水的吸附作用的分散相互作用系统中的重要性。片段和有限簇方法可以以高精度计算协议的形式结合使用,其中逐步使用较低级别的嵌入模型来评估较高级别的校正。我们应用这些技术来研究库仑筛查在磷光双层的例子和2D二氧化硅上水的吸附作用的分散相互作用系统中的重要性。

























 京公网安备 11010802027423号
京公网安备 11010802027423号