当前位置:
X-MOL 学术
›
ACS Earth Space Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Binding Geometries of Silicate Species on Ferrihydrite Surfaces
ACS Earth and Space Chemistry ( IF 3.4 ) Pub Date : 2018-01-18 00:00:00 , DOI: 10.1021/acsearthspacechem.7b00109 Xiaoming Wang 1 , James D. Kubicki 2 , Jean-François Boily 3 , Glenn A. Waychunas 4 , Yongfeng Hu 5 , Xionghan Feng 6 , Mengqiang Zhu 1
ACS Earth and Space Chemistry ( IF 3.4 ) Pub Date : 2018-01-18 00:00:00 , DOI: 10.1021/acsearthspacechem.7b00109 Xiaoming Wang 1 , James D. Kubicki 2 , Jean-François Boily 3 , Glenn A. Waychunas 4 , Yongfeng Hu 5 , Xionghan Feng 6 , Mengqiang Zhu 1
Affiliation
|
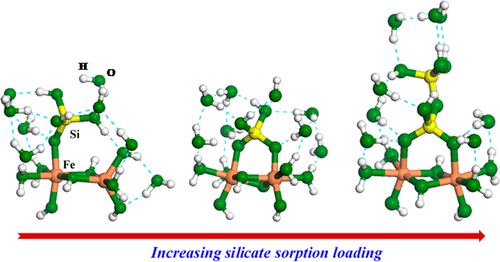
Silicate sorption on ferrihydrite surfaces, as monomers, oligomers, and polymers, strongly affects ferrihydrite crystallinity, thermodynamic stability, and surface reactivity. How these silicate species bind on ferrihydrite surfaces is, however, not well understood. We have determined silicate binding geometries using a combination of X-ray absorption spectroscopy (XAS), differential atomic pair distribution function (d-PDF) analysis, and density functional theory (DFT) calculations. Silicon K-edge absorption pre-edges and DFT-predicted energies indicate that silicate forms monomeric monodentate–mononuclear (MM) complexes at low silicate sorption loadings. With increasing silicate loading, the pre-edge peak shifts to higher energies, suggesting changes in the silicate binding geometry toward multidentate complexation. The d-PDF analysis determines the Si–Fe interatomic distance to be ∼3.25 Å for the high-loading samples. The DFT calculations indicate that such distance corresponds to an oligomer in the bidentate–binuclear (BB) binding geometry. The transition of the silicate sorption geometry accompanied by polymerization can affect stability of ferrihydrite and its adsorption and redox reactivity and increase the degree of Si isotopic fractionation upon silicate sorption on Fe oxides. MM monomeric complexes and BB oligomeric complexes should be used for surface complexation models predicting silicate sorption on Fe oxide surfaces.
中文翻译:

水铁矿表面硅酸盐物种的结合几何形状
硅酸盐作为单体,低聚物和聚合物吸附在三水铁矿表面上,会严重影响三水铁矿的结晶度,热力学稳定性和表面反应性。然而,这些硅酸盐物质如何结合在三水铁矿表面上的方法尚未广为人知。我们已经使用X射线吸收光谱(XAS),微分原子对分布函数(d-PDF)分析和密度泛函理论(DFT)计算的组合确定了硅酸盐结合的几何形状。硅K边缘吸收前缘和DFT预测的能量表明,硅酸盐在低硅酸盐吸附负荷下形成单体单齿-单核(MM)配合物。随着硅酸盐负载量的增加,前缘峰移至更高的能量,这表明硅酸盐结合几何结构朝着多齿络合的方向变化。d-PDF分析确定高负载样品的Si-Fe原子间距离为〜3.25Å。DFT计算表明,该距离对应于双齿-双核(BB)结合几何结构中的低聚物。伴随聚合的硅酸盐吸附几何形状的转变会影响三水铁矿的稳定性及其吸附和氧化还原反应性,并增加硅酸盐在Fe氧化物上吸附时Si同位素分馏的程度。MM单体配合物和BB低聚物配合物应用于表面络合模型,以预测硅酸盐在Fe氧化物表面的吸附。伴随聚合的硅酸盐吸附几何形状的转变会影响三水铁矿的稳定性及其吸附和氧化还原反应性,并增加硅酸盐在Fe氧化物上吸附时Si同位素分馏的程度。MM单体配合物和BB低聚物配合物应用于表面络合模型,以预测硅酸盐在Fe氧化物表面的吸附。伴随聚合的硅酸盐吸附几何形状的转变会影响三水铁矿的稳定性及其吸附和氧化还原反应性,并增加硅酸盐在Fe氧化物上吸附时Si同位素分馏的程度。MM单体配合物和BB低聚物配合物应用于表面络合模型,以预测硅酸盐在Fe氧化物表面的吸附。
更新日期:2018-01-18
中文翻译:
水铁矿表面硅酸盐物种的结合几何形状
硅酸盐作为单体,低聚物和聚合物吸附在三水铁矿表面上,会严重影响三水铁矿的结晶度,热力学稳定性和表面反应性。然而,这些硅酸盐物质如何结合在三水铁矿表面上的方法尚未广为人知。我们已经使用X射线吸收光谱(XAS),微分原子对分布函数(d-PDF)分析和密度泛函理论(DFT)计算的组合确定了硅酸盐结合的几何形状。硅K边缘吸收前缘和DFT预测的能量表明,硅酸盐在低硅酸盐吸附负荷下形成单体单齿-单核(MM)配合物。随着硅酸盐负载量的增加,前缘峰移至更高的能量,这表明硅酸盐结合几何结构朝着多齿络合的方向变化。d-PDF分析确定高负载样品的Si-Fe原子间距离为〜3.25Å。DFT计算表明,该距离对应于双齿-双核(BB)结合几何结构中的低聚物。伴随聚合的硅酸盐吸附几何形状的转变会影响三水铁矿的稳定性及其吸附和氧化还原反应性,并增加硅酸盐在Fe氧化物上吸附时Si同位素分馏的程度。MM单体配合物和BB低聚物配合物应用于表面络合模型,以预测硅酸盐在Fe氧化物表面的吸附。伴随聚合的硅酸盐吸附几何形状的转变会影响三水铁矿的稳定性及其吸附和氧化还原反应性,并增加硅酸盐在Fe氧化物上吸附时Si同位素分馏的程度。MM单体配合物和BB低聚物配合物应用于表面络合模型,以预测硅酸盐在Fe氧化物表面的吸附。伴随聚合的硅酸盐吸附几何形状的转变会影响三水铁矿的稳定性及其吸附和氧化还原反应性,并增加硅酸盐在Fe氧化物上吸附时Si同位素分馏的程度。MM单体配合物和BB低聚物配合物应用于表面络合模型,以预测硅酸盐在Fe氧化物表面的吸附。

























 京公网安备 11010802027423号
京公网安备 11010802027423号