Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Structural and Electronic Descriptors of Catalytic Activity of Graphene‐Based Materials: First‐Principles Theoretical Analysis
Small ( IF 13.3 ) Pub Date : 2017-12-28 , DOI: 10.1002/smll.201703609 S. Sinthika 1 , Umesh V. Waghmare 2 , Ranjit Thapa 1
Small ( IF 13.3 ) Pub Date : 2017-12-28 , DOI: 10.1002/smll.201703609 S. Sinthika 1 , Umesh V. Waghmare 2 , Ranjit Thapa 1
Affiliation
|
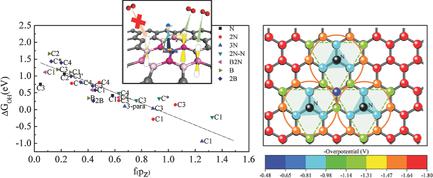
Characteristic features of the d‐band in electronic structure of transition metals are quite effective as descriptors of their catalytic activity toward oxygen reduction reaction (ORR). With the promise of graphene‐based materials to replace precious metal catalysts, descriptors of their chemical activity are much needed. Here, a site‐specific electronic descriptor is proposed based on the pz (π) orbital occupancy and its contribution to electronic states at the Fermi level. Simple structural descriptors are identified, and a linear predictive model is developed to precisely estimate adsorption free energies of OH (ΔGOH) at various sites of doped graphene, and it is demonstrated through prediction of the most optimal site for catalysis of ORR. These structural descriptors, essentially the number of ortho, meta, and para sites of N/B‐doped graphene sheet, can be extended to other doped sp2 hybridized systems, and greatly reduce the computational effort in estimating ΔGOH and site‐specific catalytic activity.
中文翻译:

石墨烯基材料催化活性的结构和电子描述符:第一原理理论分析
过渡金属电子结构中d波段的特征可以非常有效地描述其对氧还原反应(ORR)的催化活性。石墨烯基材料有望取代贵金属催化剂,因此迫切需要对其化学活性进行描述。在此,基于p z(π)轨道占有率及其对费米能级电子态的贡献,提出了一个特定于站点的电子描述符。确定简单的结构描述符,并建立线性预测模型以精确估算OH的吸附自由能(ΔG OH)在掺杂石墨烯的各个位点上,并且通过预测ORR的最佳催化位点得到了证明。这些结构描述符(本质上是N / B掺杂的石墨烯片的邻位,间位和对位)可以扩展到其他掺杂的sp 2杂化系统,并大大减少了估算ΔG OH和位点特异性的计算量催化活性。
更新日期:2017-12-28
中文翻译:
石墨烯基材料催化活性的结构和电子描述符:第一原理理论分析
过渡金属电子结构中d波段的特征可以非常有效地描述其对氧还原反应(ORR)的催化活性。石墨烯基材料有望取代贵金属催化剂,因此迫切需要对其化学活性进行描述。在此,基于p z(π)轨道占有率及其对费米能级电子态的贡献,提出了一个特定于站点的电子描述符。确定简单的结构描述符,并建立线性预测模型以精确估算OH的吸附自由能(ΔG OH)在掺杂石墨烯的各个位点上,并且通过预测ORR的最佳催化位点得到了证明。这些结构描述符(本质上是N / B掺杂的石墨烯片的邻位,间位和对位)可以扩展到其他掺杂的sp 2杂化系统,并大大减少了估算ΔG OH和位点特异性的计算量催化活性。
























 京公网安备 11010802027423号
京公网安备 11010802027423号