当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Reverse and Random Decoy Methods for False Discovery Rate Estimation in High Mass Accuracy Peptide Spectral Library Searches
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2018-01-11 00:00:00 , DOI: 10.1021/acs.jproteome.7b00614 Zheng Zhang 1 , Meghan Burke 1 , Yuri A. Mirokhin 1 , Dmitrii V. Tchekhovskoi 1 , Sanford P. Markey 1 , Wen Yu 2 , Raghothama Chaerkady 3 , Sonja Hess 3 , Stephen E. Stein 1
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2018-01-11 00:00:00 , DOI: 10.1021/acs.jproteome.7b00614 Zheng Zhang 1 , Meghan Burke 1 , Yuri A. Mirokhin 1 , Dmitrii V. Tchekhovskoi 1 , Sanford P. Markey 1 , Wen Yu 2 , Raghothama Chaerkady 3 , Sonja Hess 3 , Stephen E. Stein 1
Affiliation
|
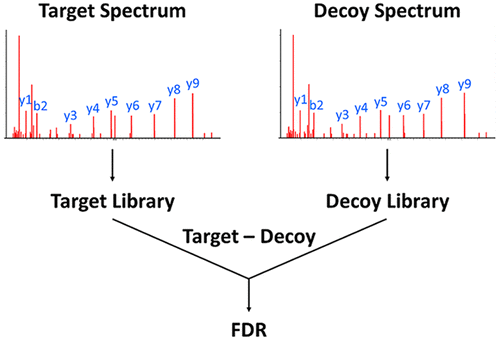
Spectral library searching (SLS) is an attractive alternative to sequence database searching (SDS) for peptide identification due to its speed, sensitivity, and ability to include any selected mass spectra. While decoy methods for SLS have been developed for low mass accuracy peptide spectral libraries, it is not clear that they are optimal or directly applicable to high mass accuracy spectra. Therefore, we report the development and validation of methods for high mass accuracy decoy libraries. Two types of decoy libraries were found to be suitable for this purpose. The first, referred to as Reverse, constructs spectra by reversing a library’s peptide sequences except for the C-terminal residue. The second, termed Random, randomly replaces all non-C-terminal residues and either retains the original C-terminal residue or replaces it based on the amino-acid frequency of the library’s C-terminus. In both cases the m/z values of fragment ions are shifted accordingly. Determination of FDR is performed in a manner equivalent to SDS, concatenating a library with its decoy prior to a search. The utility of Reverse and Random libraries for target-decoy SLS in estimating false-positives and FDRs was demonstrated using spectra derived from a recently published synthetic human proteome project (Zolg, D. P.; et al. Nat. Methods 2017, 14, 259–262). For data sets from two large-scale label-free and iTRAQ experiments, these decoy building methods yielded highly similar score thresholds and spectral identifications at 1% FDR. The results were also found to be equivalent to those of using the decoy-free PeptideProphet algorithm. Using these new methods for FDR estimation, MSPepSearch, which is freely available search software, led to 18% more identifications at 1% FDR and 23% more at 0.1% FDR when compared with other widely used SDS engines coupled to postprocessing approaches such as Percolator. An application of these methods for FDR estimation for the recently reported “hybrid” library search (Burke, M. C.; et al. J. Proteome Res. 2017, 16, 1924–1935) method is also made. The application of decoy methods for high mass accuracy SLS permits the merging of these results with those of SDS, thereby increasing the assignment of more peptides, leading to deeper proteome coverage.
中文翻译:

反向和随机诱饵方法在高质量肽谱库检索中错误发现率估计中的应用
光谱库搜索(SLS)是用于肽段鉴定的序列数据库搜索(SDS)的一种有吸引力的替代方法,因为它的速度,灵敏度以及包括任何选定质谱图的能力。尽管针对低质量精度的肽谱库开发了SLS诱饵方法,但尚不清楚它们是最佳方法还是直接适用于高质量精度的谱图。因此,我们报告了高质量诱饵库的开发方法和验证。发现两种类型的诱饵库适合于此目的。第一个称为“反向”,通过反转库的C端残基以外的肽序列来构建光谱。第二种称为随机 随机替换所有非C末端残基,并保留原始C末端残基或根据文库C末端的氨基酸频率替换它。在两种情况下碎片离子的m / z值会相应移动。FDR的确定以与SDS等效的方式执行,在搜索之前将库及其诱饵连接起来。。反向和随机文库在估计假阳性和一级亲属使用来自最近发表的合成的人类蛋白质组计划衍生(Zolg,DP谱证实靶诱饵SLS的效用;等。纳特方法 2017,14 ,259–262)。对于来自两个大规模无标签实验和iTRAQ实验的数据集,这些诱饵构建方法在FDR为1%时产生了非常相似的得分阈值和光谱鉴定。还发现结果与使用无诱饵的PeptideProphet算法的结果相同。与其他广泛使用的SDS引擎(例如Percolator)相比,使用这些新方法进行FDR估算时,MSPepSearch(可免费获得的搜索软件)在1%FDR时的识别率提高了18%,在0.1%FDR时的识别率提高了23%。 。这些方法对于FDR估计最近报道的“混合”的应用程序库搜索(伯克,MC;等人,J.蛋白质组RES。 2017年,16,1924-1935年)的方法。诱饵方法用于高质量SLS的应用允许将这些结果与SDS的结果合并,从而增加更多肽的分配,从而实现更深的蛋白质组覆盖。
更新日期:2018-01-11
中文翻译:
反向和随机诱饵方法在高质量肽谱库检索中错误发现率估计中的应用
光谱库搜索(SLS)是用于肽段鉴定的序列数据库搜索(SDS)的一种有吸引力的替代方法,因为它的速度,灵敏度以及包括任何选定质谱图的能力。尽管针对低质量精度的肽谱库开发了SLS诱饵方法,但尚不清楚它们是最佳方法还是直接适用于高质量精度的谱图。因此,我们报告了高质量诱饵库的开发方法和验证。发现两种类型的诱饵库适合于此目的。第一个称为“反向”,通过反转库的C端残基以外的肽序列来构建光谱。第二种称为随机 随机替换所有非C末端残基,并保留原始C末端残基或根据文库C末端的氨基酸频率替换它。在两种情况下碎片离子的m / z值会相应移动。FDR的确定以与SDS等效的方式执行,在搜索之前将库及其诱饵连接起来。。反向和随机文库在估计假阳性和一级亲属使用来自最近发表的合成的人类蛋白质组计划衍生(Zolg,DP谱证实靶诱饵SLS的效用;等。纳特方法 2017,14 ,259–262)。对于来自两个大规模无标签实验和iTRAQ实验的数据集,这些诱饵构建方法在FDR为1%时产生了非常相似的得分阈值和光谱鉴定。还发现结果与使用无诱饵的PeptideProphet算法的结果相同。与其他广泛使用的SDS引擎(例如Percolator)相比,使用这些新方法进行FDR估算时,MSPepSearch(可免费获得的搜索软件)在1%FDR时的识别率提高了18%,在0.1%FDR时的识别率提高了23%。 。这些方法对于FDR估计最近报道的“混合”的应用程序库搜索(伯克,MC;等人,J.蛋白质组RES。 2017年,16,1924-1935年)的方法。诱饵方法用于高质量SLS的应用允许将这些结果与SDS的结果合并,从而增加更多肽的分配,从而实现更深的蛋白质组覆盖。


























 京公网安备 11010802027423号
京公网安备 11010802027423号