当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
The 3D Structure of Human DP Prostaglandin G-Protein-Coupled Receptor Bound to Cyclopentanoindole Antagonist, Predicted Using the DuplexBiHelix Modification of the GEnSeMBLE Method
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2017-12-21 00:00:00 , DOI: 10.1021/acs.jctc.7b00842 Vishnu Shankar 1 , William A. Goddard 1 , Soo-Kyung Kim 1 , Ravinder Abrol 1 , Fan Liu 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2017-12-21 00:00:00 , DOI: 10.1021/acs.jctc.7b00842 Vishnu Shankar 1 , William A. Goddard 1 , Soo-Kyung Kim 1 , Ravinder Abrol 1 , Fan Liu 1
Affiliation
|
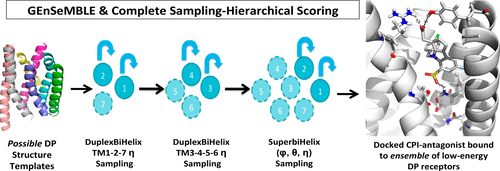
Prostaglandins play a critical physiological role in both cardiovascular and immune systems, acting through their interactions with 9 prostanoid G protein-coupled receptors (GPCRs). These receptors are important therapeutic targets for a variety of diseases including arthritis, allergies, type 2 diabetes, and cancer. The DP prostaglandin receptor is of interest because it has unique structural and physiological properties. Most notably, DP does not have the 3–6 ionic lock common to Class A GPCRs. However, the lack of X-ray structures for any of the 9 prostaglandin GPCRs hampers the application of structure-based drug design methods to develop more selective and active medications to specific receptors. We predict here 3D structures for the DP prostaglandin GPCR, based on the GEnSeMBLE complete sampling with hierarchical scoring (CS-HS) methodology. This involves evaluating the energy of 13 trillion packings to finally select the best 20 that are stable enough to be relevant for binding to antagonists, agonists, and modulators. To validate the predicted structures, we predict the binding site for the Merck cyclopentanoindole (CPI) selective antagonist docked to DP. We find that the CPI binds vertically in the 1–2–7 binding pocket, interacting favorably with residues R3107.40 and K762.54 with additional interactions with S3137.43, S3167.46, S191.35, etc. This binding site differs significantly from that of antagonists to known Class A GPCRs where the ligand binds in the 3–4–5–6 region. We find that the predicted binding site leads to reasonable agreement with experimental Structure–Activity Relationship (SAR). We suggest additional mutation experiments including K762.54, E1293.49, L1233.43, M2706.40, F2746.44 to further validate the structure, function, and activation mechanism of receptors in the prostaglandin family. Our structures and binding sites are largely consistent and improve upon the predictions by Li et al. ( J. Am. Chem. Soc. 2007, 129 (35), 10720) that used our earlier MembStruk prediction methodology.
中文翻译:

使用DPnBiHelix修饰的GEnSeMBLE方法预测人DP前列腺素G蛋白偶联受体与环戊烷吲哚拮抗剂的3D结构
前列腺素通过与9种前列腺素G蛋白偶联受体(GPCR)相互作用,在心血管和免疫系统中均起着至关重要的生理作用。这些受体是多种疾病(包括关节炎,过敏,2型糖尿病和癌症)的重要治疗靶标。DP前列腺素受体是令人感兴趣的,因为它具有独特的结构和生理特性。最值得注意的是,DP没有A类GPCR共有的3–6离子锁。但是,对于9种前列腺素GPCR中的任何一种,X射线结构的缺乏都阻碍了基于结构的药物设计方法的应用,从而无法开发出针对特定受体的更具选择性和活性的药物。我们在此基于具有分层评分(CS-HS)方法的GEnSeMBLE完整采样,预测了DP前列腺素GPCR的3D结构。这涉及评估13万亿个填充物的能量,以最终选择最好的20种填充物,这些填充物足够稳定,足以与拮抗剂,激动剂和调节剂结合。为了验证预测的结构,我们预测了停泊在DP上的Merck环戊吲哚(CPI)选择性拮抗剂的结合位点。我们发现CPI在1–2–7的结合袋中垂直结合,与残基R310相互作用良好7.40和K76 2.54以及与S313 7.43,S316 7.46,S19 1.35等的其他相互作用。该结合位点与已知A类GPCR拮抗剂的结合位点显着不同,后者的配体在3–4–5–6区域结合。我们发现预测的结合位点导致与实验的结构-活性关系(SAR)达成合理的一致。我们建议进行其他突变实验,包括K76 2.54,E129 3.49,L123 3.43,M270 6.40,F274 6.44进一步验证前列腺素家族受体的结构,功能和激活机制。我们的结构和结合位点在很大程度上是一致的,并根据Li等人的预测进行了改进。(J.化学会会志。 2007年,129(35),10720),其用于我们的早期MembStruk预测方法。
更新日期:2017-12-21
中文翻译:
使用DPnBiHelix修饰的GEnSeMBLE方法预测人DP前列腺素G蛋白偶联受体与环戊烷吲哚拮抗剂的3D结构
前列腺素通过与9种前列腺素G蛋白偶联受体(GPCR)相互作用,在心血管和免疫系统中均起着至关重要的生理作用。这些受体是多种疾病(包括关节炎,过敏,2型糖尿病和癌症)的重要治疗靶标。DP前列腺素受体是令人感兴趣的,因为它具有独特的结构和生理特性。最值得注意的是,DP没有A类GPCR共有的3–6离子锁。但是,对于9种前列腺素GPCR中的任何一种,X射线结构的缺乏都阻碍了基于结构的药物设计方法的应用,从而无法开发出针对特定受体的更具选择性和活性的药物。我们在此基于具有分层评分(CS-HS)方法的GEnSeMBLE完整采样,预测了DP前列腺素GPCR的3D结构。这涉及评估13万亿个填充物的能量,以最终选择最好的20种填充物,这些填充物足够稳定,足以与拮抗剂,激动剂和调节剂结合。为了验证预测的结构,我们预测了停泊在DP上的Merck环戊吲哚(CPI)选择性拮抗剂的结合位点。我们发现CPI在1–2–7的结合袋中垂直结合,与残基R310相互作用良好7.40和K76 2.54以及与S313 7.43,S316 7.46,S19 1.35等的其他相互作用。该结合位点与已知A类GPCR拮抗剂的结合位点显着不同,后者的配体在3–4–5–6区域结合。我们发现预测的结合位点导致与实验的结构-活性关系(SAR)达成合理的一致。我们建议进行其他突变实验,包括K76 2.54,E129 3.49,L123 3.43,M270 6.40,F274 6.44进一步验证前列腺素家族受体的结构,功能和激活机制。我们的结构和结合位点在很大程度上是一致的,并根据Li等人的预测进行了改进。(J.化学会会志。 2007年,129(35),10720),其用于我们的早期MembStruk预测方法。

























 京公网安备 11010802027423号
京公网安备 11010802027423号