当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Development of Embedded and Performance of Density Functional Methods for Molecular Crystals
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2018-01-05 00:00:00 , DOI: 10.1021/acs.jpca.7b12467 Grygoriy A. Dolgonos 1 , Oleksandr A. Loboda 1 , A. Daniel Boese 1
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2018-01-05 00:00:00 , DOI: 10.1021/acs.jpca.7b12467 Grygoriy A. Dolgonos 1 , Oleksandr A. Loboda 1 , A. Daniel Boese 1
Affiliation
|
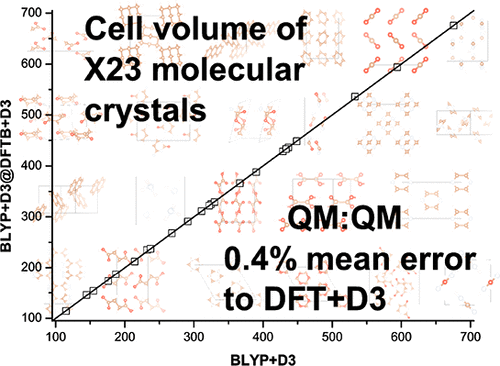
We report an alternative quantum mechanical:quantum mechanical (QM:QM) method to the currently used periodic density functional calculations including dispersion and investigate its performance with respect to main structural and energetic properties of the X23 set of molecular crystals. By setting the goal of reproducing reference periodic BLYP+D3 values and by embedding BLYP+D3 into DFTB, we obtain results similar to those of periodic BLYP+D3—typically within 1–2% in lattice energies and ∼0.4% in cell volumes. The accuracy of this QM:QM method in comparison to DFTB+D and DFT+D for the X23 set of molecular crystals is discussed.
中文翻译:

分子晶体的嵌入和密度泛函方法的发展
我们报告了一种替代的量子力学:量子力学(QM:QM)方法,用于当前使用的周期性密度泛函计算,包括色散,并针对分子晶体X23集的主要结构和高能性质研究其性能。通过设定复制参考周期性BLYP + D3值并将BLYP + D3嵌入DFTB的目标,我们获得了与周期性BLYP + D3相似的结果-通常在1-2%的晶格能量和〜0.4%的晶胞能量内。讨论了与XTB组分子晶体的DFTB + D和DFT + D相比,此QM:QM方法的准确性。
更新日期:2018-01-05
中文翻译:
分子晶体的嵌入和密度泛函方法的发展
我们报告了一种替代的量子力学:量子力学(QM:QM)方法,用于当前使用的周期性密度泛函计算,包括色散,并针对分子晶体X23集的主要结构和高能性质研究其性能。通过设定复制参考周期性BLYP + D3值并将BLYP + D3嵌入DFTB的目标,我们获得了与周期性BLYP + D3相似的结果-通常在1-2%的晶格能量和〜0.4%的晶胞能量内。讨论了与XTB组分子晶体的DFTB + D和DFT + D相比,此QM:QM方法的准确性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号