当前位置:
X-MOL 学术
›
Biochemistry
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Where Informatics Lags Chemistry Leads
Biochemistry ( IF 2.9 ) Pub Date : 2017-12-22 00:00:00 , DOI: 10.1021/acs.biochem.7b01073 Rahul Kaushik 1, 2 , Ankita Singh 2, 3 , B. Jayaram 1, 2, 4
Biochemistry ( IF 2.9 ) Pub Date : 2017-12-22 00:00:00 , DOI: 10.1021/acs.biochem.7b01073 Rahul Kaushik 1, 2 , Ankita Singh 2, 3 , B. Jayaram 1, 2, 4
Affiliation
|
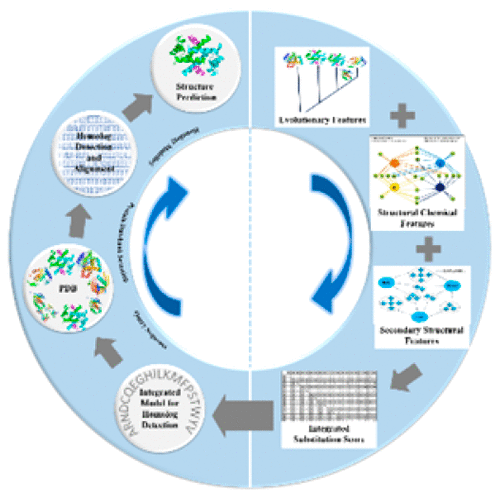
The fact that amino acid sequences dictate the tertiary structures of proteins has been known for more than five decades. While the molecular pathways to tertiary structure are still being worked out, with the axiom that similar sequences adopt similar structures, computational methods are being developed continually in parallel, utilizing the Protein Data Bank structural repository and homologue detection strategies to predict structures of sequences of interest. The success of this approach is limited by the ability to unravel the hidden similarities among amino acid sequences. We consider here the 20 amino acids as a complete set of chemical templates in the physicochemical space of proteins and propose a new structural and chemical classification of amino acids. An integration of this perspective into the conventional evolutionary methods of similarity detection leads to an unprecedented increase in the accuracy in homologue detection, resulting in improved protein structure prediction. The performance is validated on a large data set of 11716 unique proteins, and the results are benchmarked against conventional methods. The availability of good quality protein structures helps in structure-based drug design endeavors and in establishing protein structure–function correlations.
中文翻译:

信息学落后化学的地方
氨基酸序列决定蛋白质的三级结构的事实已超过五十年了。尽管仍在研究通往三级结构的分子途径,但公理是相似的序列采用相似的结构,但是并行不断开发计算方法,利用蛋白质数据库的结构库和同源物检测策略来预测目标序列的结构。该方法的成功受到解开氨基酸序列之间隐藏的相似性的能力的限制。我们在这里将20个氨基酸视为蛋白质物理化学空间中的完整化学模板集,并提出了氨基酸的新结构和化学分类。将这种观点整合到相似性检测的传统进化方法中,导致同源检测准确性的空前提高,从而改善了蛋白质结构的预测。该性能已在11716种独特蛋白质的大型数据集上得到验证,并且结果与常规方法进行了比较。高质量蛋白质结构的可用性有助于进行基于结构的药物设计,并有助于建立蛋白质结构与功能的相关性。
更新日期:2017-12-22
中文翻译:
信息学落后化学的地方
氨基酸序列决定蛋白质的三级结构的事实已超过五十年了。尽管仍在研究通往三级结构的分子途径,但公理是相似的序列采用相似的结构,但是并行不断开发计算方法,利用蛋白质数据库的结构库和同源物检测策略来预测目标序列的结构。该方法的成功受到解开氨基酸序列之间隐藏的相似性的能力的限制。我们在这里将20个氨基酸视为蛋白质物理化学空间中的完整化学模板集,并提出了氨基酸的新结构和化学分类。将这种观点整合到相似性检测的传统进化方法中,导致同源检测准确性的空前提高,从而改善了蛋白质结构的预测。该性能已在11716种独特蛋白质的大型数据集上得到验证,并且结果与常规方法进行了比较。高质量蛋白质结构的可用性有助于进行基于结构的药物设计,并有助于建立蛋白质结构与功能的相关性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号