当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Refining Crystal Structures with Quadrupolar NMR and Dispersion-Corrected Density Functional Theory
The Journal of Physical Chemistry C ( IF 3.7 ) Pub Date : 2018-01-16 00:00:00 , DOI: 10.1021/acs.jpcc.7b12314 Sean T. Holmes 1 , Robert W. Schurko 1
The Journal of Physical Chemistry C ( IF 3.7 ) Pub Date : 2018-01-16 00:00:00 , DOI: 10.1021/acs.jpcc.7b12314 Sean T. Holmes 1 , Robert W. Schurko 1
Affiliation
|
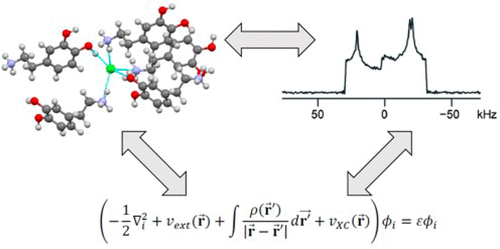
Nuclear electric field gradient (EFG) tensors obtained from solid-state NMR spectroscopy are highly responsive to variations in structural features. The orientations and principal components of EFG tensors show great variation between different molecular structures; hence, extraction of EFG tensor parameters, either experimentally or computationally, provides a powerful means for structure determination and refinement. Here, dispersion-corrected plane-wave density functional theory (DFT) is used to refine atomic coordinates in organic crystals determined initially through single-crystal X-ray diffraction (XRD) or neutron diffraction methods. To accomplish this, an empirical parametrization of a two-body dispersion force field is illustrated, in which comparisons of experimental and calculated 14N, 17O, and 35Cl EFG tensor parameters are used to assess the quality of energy-minimized structures. The parametrization is based on a training set of 17 organic solids. The analysis is applied subsequently to the structural refinements of structural models from over 60 different materials. For the prediction of 35Cl EFG tensor parameters in particular, the optimization protocols described herein lead to a substantial improvement in agreement with experiment relative to structures obtained by XRD methods or by refinement with plane-wave DFT without the inclusion of the force field. The results further demonstrate that crystal structures with atomic coordinates refined with the present methods are able to pinpoint the positions of hydrogen atoms participating in H···Cl– hydrogen bonding with a higher degree of precision than is possible through neutron diffraction. This methodology, which is facile to implement within most DFT software packages, should prove to be very useful for future structural refinements using NMR crystallographic methods.
中文翻译:

使用四极核磁共振和色散校正的密度泛函理论精炼晶体结构
从固态NMR光谱学获得的核电场梯度(EFG)张量对结构特征的变化高度敏感。EFG张量的取向和主要成分在不同的分子结构之间显示出很大的差异。因此,通过实验或计算方法提取EFG张量参数,为确定和改进结构提供了有力的手段。在这里,使用色散校正的平面波密度泛函理论(DFT)来细化最初通过单晶X射线衍射(XRD)或中子衍射方法确定的有机晶体中的原子坐标。为此,说明了两体色散力场的经验参数化,其中比较了实验值和计算值14 N,17 O和35 Cl EFG张量参数用于评估能量最小化结构的质量。参数化基于17种有机固体的训练集。随后将分析应用于从60多种不同材料中进行的结构模型的结构优化。特别是对于35 Cl EFG张量参数的预测,相对于通过XRD方法获得的结构,或通过不包含力场的平面波DFT精修而成,本文所述的优化协议可带来与实验相一致的显着改进。结果进一步证明,用本方法精制的具有原子坐标的晶体结构能够查明参与H···Cl的氢原子的位置。–氢键具有比中子衍射更高的精确度。这种方法易于在大多数DFT软件包中实施,对于使用NMR晶体学方法进行的将来的结构改进,应该被证明是非常有用的。
更新日期:2018-01-16
中文翻译:
使用四极核磁共振和色散校正的密度泛函理论精炼晶体结构
从固态NMR光谱学获得的核电场梯度(EFG)张量对结构特征的变化高度敏感。EFG张量的取向和主要成分在不同的分子结构之间显示出很大的差异。因此,通过实验或计算方法提取EFG张量参数,为确定和改进结构提供了有力的手段。在这里,使用色散校正的平面波密度泛函理论(DFT)来细化最初通过单晶X射线衍射(XRD)或中子衍射方法确定的有机晶体中的原子坐标。为此,说明了两体色散力场的经验参数化,其中比较了实验值和计算值14 N,17 O和35 Cl EFG张量参数用于评估能量最小化结构的质量。参数化基于17种有机固体的训练集。随后将分析应用于从60多种不同材料中进行的结构模型的结构优化。特别是对于35 Cl EFG张量参数的预测,相对于通过XRD方法获得的结构,或通过不包含力场的平面波DFT精修而成,本文所述的优化协议可带来与实验相一致的显着改进。结果进一步证明,用本方法精制的具有原子坐标的晶体结构能够查明参与H···Cl的氢原子的位置。–氢键具有比中子衍射更高的精确度。这种方法易于在大多数DFT软件包中实施,对于使用NMR晶体学方法进行的将来的结构改进,应该被证明是非常有用的。
























 京公网安备 11010802027423号
京公网安备 11010802027423号