当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Constructing Grids for Molecular Quantum Dynamics Using an Autoencoder
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2017-12-27 00:00:00 , DOI: 10.1021/acs.jctc.7b01045 Julius P. P. Zauleck 1 , Regina de Vivie-Riedle 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2017-12-27 00:00:00 , DOI: 10.1021/acs.jctc.7b01045 Julius P. P. Zauleck 1 , Regina de Vivie-Riedle 1
Affiliation
|
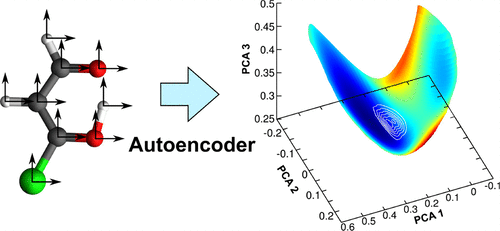
A challenge for molecular quantum dynamics (QD) calculations is the curse of dimensionality with respect to the nuclear degrees of freedom. A common approach that works especially well for fast reactive processes is to reduce the dimensionality of the system to a few most relevant coordinates. Identifying these can become a very difficult task, because they often are highly unintuitive. We present a machine learning approach that utilizes an autoencoder that is trained to find a low-dimensional representation of a set of molecular configurations. These configurations are generated by trajectory calculations performed on the reactive molecular systems of interest. The resulting low-dimensional representation can be used to generate a potential energy surface grid in the desired subspace. Using the G-matrix formalism to calculate the kinetic energy operator, QD calculations can be carried out on this grid. In addition to step-by-step instructions for the grid construction, we present the application to a test system.
中文翻译:

使用自动编码器为分子量子动力学构建网格
分子量子动力学(QD)计算面临的挑战是相对于核自由度的维数诅咒。对于快速反应过程特别有效的常见方法是将系统的维数减少到几个最相关的坐标。识别这些内容可能会变得非常困难,因为它们通常非常不直观。我们提出了一种机器学习方法,该方法利用经过训练的自动编码器来查找一组分子构型的低维表示。这些构型是通过对目标反应性分子系统进行轨迹计算而生成的。所得的低维表示可用于在所需子空间中生成势能表面网格。使用G-矩阵形式主义来计算动能算子,可以在此网格上执行QD计算。除了有关网格构建的分步说明之外,我们还将应用程序介绍给测试系统。
更新日期:2017-12-27
中文翻译:
使用自动编码器为分子量子动力学构建网格
分子量子动力学(QD)计算面临的挑战是相对于核自由度的维数诅咒。对于快速反应过程特别有效的常见方法是将系统的维数减少到几个最相关的坐标。识别这些内容可能会变得非常困难,因为它们通常非常不直观。我们提出了一种机器学习方法,该方法利用经过训练的自动编码器来查找一组分子构型的低维表示。这些构型是通过对目标反应性分子系统进行轨迹计算而生成的。所得的低维表示可用于在所需子空间中生成势能表面网格。使用G-矩阵形式主义来计算动能算子,可以在此网格上执行QD计算。除了有关网格构建的分步说明之外,我们还将应用程序介绍给测试系统。


























 京公网安备 11010802027423号
京公网安备 11010802027423号