当前位置:
X-MOL 学术
›
Chem. Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Probing Solid–Solid Interfacial Reactions in All-Solid-State Sodium-Ion Batteries with First-Principles Calculations
Chemistry of Materials ( IF 8.6 ) Pub Date : 2017-12-28 00:00:00 , DOI: 10.1021/acs.chemmater.7b04096 Hanmei Tang 1 , Zhi Deng 1 , Zhuonan Lin 2 , Zhenbin Wang 1 , Iek-Heng Chu 1 , Chi Chen 1 , Zhuoying Zhu 1 , Chen Zheng 1 , Shyue Ping Ong 1
Chemistry of Materials ( IF 8.6 ) Pub Date : 2017-12-28 00:00:00 , DOI: 10.1021/acs.chemmater.7b04096 Hanmei Tang 1 , Zhi Deng 1 , Zhuonan Lin 2 , Zhenbin Wang 1 , Iek-Heng Chu 1 , Chi Chen 1 , Zhuoying Zhu 1 , Chen Zheng 1 , Shyue Ping Ong 1
Affiliation
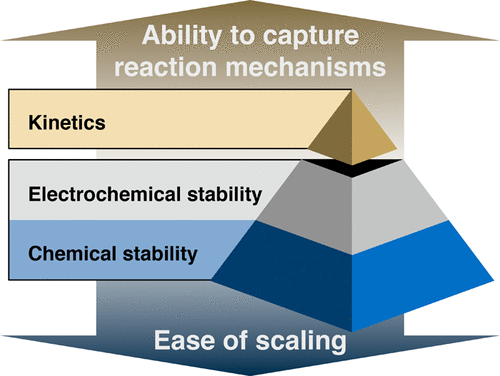
|
We present an exposition of first-principles approaches to elucidating interfacial reactions in all-solid-state sodium-ion batteries. We will demonstrate how thermodynamic approximations based on assumptions of fast alkali diffusion and multispecies equilibrium can be used to effectively screen combinations of Na-ion electrodes, solid electrolytes, and buffer oxides for electrochemical and chemical compatibility. We find that exchange reactions, especially between simple oxides and thiophosphate groups to form PO43–, are the main cause of large driving forces for cathode/solid electrolyte interfacial reactions. A high reactivity with large volume changes is also predicted at the Na anode/solid electrolyte interface, while the Na2Ti3O7 anode is predicted to be much more stable against a broad range of solid electrolytes. We identify several promising binary oxides, Sc2O3, SiO2, TiO2, ZrO2, and HfO2, that are similarly or more chemically compatible with most electrodes and solid electrolytes than the commonly used Al2O3 is. Finally, we show that ab initio molecular dynamics simulations of the NaCoO2/Na3PS4 interface model predict that the formation of SO42–-containing compounds and Na3P is kinetically favored over the formation of PO43–-containing compounds, in contrast to the predictions of the thermodynamic models. This work provides useful insights into materials selection strategies for enabling stable electrode/solid electrolyte interfaces, a critical bottleneck in designing all-solid-state sodium-ion batteries, and outlines several testable predictions for future experimental validation.
中文翻译:

用第一性原理探索全固态钠离子电池中的固-固界面反应
我们提出了阐明所有固态钠离子电池中界面反应的第一性原理方法的说明。我们将演示如何基于快速碱扩散和多物种平衡的假设进行热力学近似,以有效筛选Na离子电极,固体电解质和缓冲氧化物的组合,以实现电化学和化学相容性。我们发现交换反应,特别是在简单的氧化物和硫代磷酸盐基团之间形成PO 4 3–的交换反应,是阴极/固体电解质界面反应产生较大驱动力的主要原因。在Na阳极/固体电解质界面处,还预测到高反应性和大体积变化,而Na 2 Ti 3 O预计7阳极对广泛的固体电解质更稳定。我们确定了几种有前途的二元氧化物Sc 2 O 3,SiO 2,TiO 2,ZrO 2和HfO 2,与大多数电极和固体电解质相比,化学或化学相容性高于常用的Al 2 O 3。最后,我们证明了NaCoO 2 / Na 3 PS 4界面模型的从头算分子动力学模拟可以预测含SO 4 2的化合物和Na 3的形成。与热力学模型的预测相反,P在动力学上优于含PO 4 3–的化合物的形成。这项工作为实现稳定的电极/固体电解质界面的材料选择策略提供了有用的见识,这是设计全固态钠离子电池的关键瓶颈,并概述了一些可测试的预测,以供将来进行实验验证。
更新日期:2017-12-28
中文翻译:
用第一性原理探索全固态钠离子电池中的固-固界面反应
我们提出了阐明所有固态钠离子电池中界面反应的第一性原理方法的说明。我们将演示如何基于快速碱扩散和多物种平衡的假设进行热力学近似,以有效筛选Na离子电极,固体电解质和缓冲氧化物的组合,以实现电化学和化学相容性。我们发现交换反应,特别是在简单的氧化物和硫代磷酸盐基团之间形成PO 4 3–的交换反应,是阴极/固体电解质界面反应产生较大驱动力的主要原因。在Na阳极/固体电解质界面处,还预测到高反应性和大体积变化,而Na 2 Ti 3 O预计7阳极对广泛的固体电解质更稳定。我们确定了几种有前途的二元氧化物Sc 2 O 3,SiO 2,TiO 2,ZrO 2和HfO 2,与大多数电极和固体电解质相比,化学或化学相容性高于常用的Al 2 O 3。最后,我们证明了NaCoO 2 / Na 3 PS 4界面模型的从头算分子动力学模拟可以预测含SO 4 2的化合物和Na 3的形成。与热力学模型的预测相反,P在动力学上优于含PO 4 3–的化合物的形成。这项工作为实现稳定的电极/固体电解质界面的材料选择策略提供了有用的见识,这是设计全固态钠离子电池的关键瓶颈,并概述了一些可测试的预测,以供将来进行实验验证。


























 京公网安备 11010802027423号
京公网安备 11010802027423号