当前位置:
X-MOL 学术
›
Mol. Pharmaceutics
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Inhibition of Human CYP3A4 by Rationally Designed Ritonavir-Like Compounds: Impact and Interplay of the Side Group Functionalities
Molecular Pharmaceutics ( IF 4.9 ) Pub Date : 2017-12-12 00:00:00 , DOI: 10.1021/acs.molpharmaceut.7b00957 Eric R. Samuels 1 , Irina Sevrioukova 1
Molecular Pharmaceutics ( IF 4.9 ) Pub Date : 2017-12-12 00:00:00 , DOI: 10.1021/acs.molpharmaceut.7b00957 Eric R. Samuels 1 , Irina Sevrioukova 1
Affiliation
|
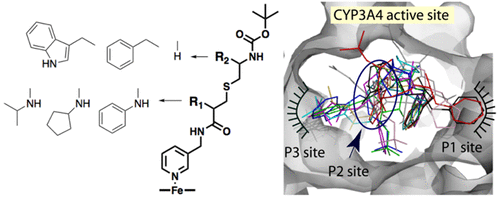
Structure–function relationships of nine rationally designed ritonavir-like compounds were investigated to better understand the ligand binding and inhibitory mechanism in human drug-metabolizing cytochrome P450 3A4 (CYP3A4). The analogs had a similar backbone and pyridine and tert-butyloxycarbonyl (Boc) as the heme-ligating and terminal groups, respectively. N-Isopropyl, N-cyclopentyl, or N-phenyl were the R1-side group substituents alone (compounds 5a–c) or in combination with phenyl or indole at the R2 position (8a–c and 8d–f subseries, respectively). Our experimental and structural data indicate that (i) for all analogs, a decrease in the dissociation constant (Ks) coincides with a decrease in IC50, but no relation with other derived parameters is observed; (ii) an increase in the R1 volume, hydrophobicity, and aromaticity markedly lowers Ks and IC50, whereas the addition of aromatic R2 has a more pronounced positive effect on the inhibitory potency than the binding strength; (iii) the ligands’ association mode is strongly influenced by the mutually dependent R1–R2 interplay, but the R1-mediated interactions are dominant and define the overall conformation in the active site; (iv) formation of a strong H-bond with Ser119 is a prerequisite for potent CYP3A4 inhibition; and (v) the strongest inhibitor in the series, the R1-phenyl/R2-indole containing 8f (Ks and IC50 of 0.08 and 0.43 μM, respectively), is still less potent than ritonavir, even under conditions that prevent the mechanism based inactivation of CYP3A4. Crystallographic data were essential for better understanding and interpretation of the experimental results, and suggested how the inhibitor design could be further optimized.
中文翻译:

合理设计的利托那韦样化合物对人CYP3A4的抑制作用:侧基功能的影响和相互作用
研究了九种合理设计的利托那韦样化合物的结构与功能之间的关系,以更好地了解人类药物代谢细胞色素P450 3A4(CYP3A4)中的配体结合和抑制机制。类似物具有与血红素连接基团和末端基团相似的主链,吡啶和叔丁氧羰基(Boc)。N-异丙基,N-环戊基或N-苯基是单独的R 1侧基取代基(化合物5a – c),或与R 2位置上的苯基或吲哚结合(8a – c和8d – f)子系列)。我们的实验和结构数据表明:(i)对于所有类似物,解离常数(K s)的降低与IC 50的降低相吻合,但未观察到与其他衍生参数的关系;(ii)R 1体积,疏水性和芳族性的增加显着降低了K s和IC 50,而芳族R 2的添加对抑制力的显着积极作用大于其结合强度;(iii)配体的缔合模式受到相互依赖的R 1 -R的强烈影响2种相互作用,但R 1介导的相互作用占主导地位,并定义了活性位点的整体构象;(iv)与Ser119形成强H键是有效抑制CYP3A4的先决条件; 和(v)系列中最强的抑制剂,R 1-苯基/ R 2-吲哚含8f(K s和IC 50即使在阻止基于CYP3A4的机制失活的条件下,其分别为0.08和0.43μM的浓度仍然比利托那韦的效力低。晶体学数据对于更好地理解和解释实验结果至关重要,并建议了如何进一步优化抑制剂的设计。
更新日期:2017-12-12
中文翻译:
合理设计的利托那韦样化合物对人CYP3A4的抑制作用:侧基功能的影响和相互作用
研究了九种合理设计的利托那韦样化合物的结构与功能之间的关系,以更好地了解人类药物代谢细胞色素P450 3A4(CYP3A4)中的配体结合和抑制机制。类似物具有与血红素连接基团和末端基团相似的主链,吡啶和叔丁氧羰基(Boc)。N-异丙基,N-环戊基或N-苯基是单独的R 1侧基取代基(化合物5a – c),或与R 2位置上的苯基或吲哚结合(8a – c和8d – f)子系列)。我们的实验和结构数据表明:(i)对于所有类似物,解离常数(K s)的降低与IC 50的降低相吻合,但未观察到与其他衍生参数的关系;(ii)R 1体积,疏水性和芳族性的增加显着降低了K s和IC 50,而芳族R 2的添加对抑制力的显着积极作用大于其结合强度;(iii)配体的缔合模式受到相互依赖的R 1 -R的强烈影响2种相互作用,但R 1介导的相互作用占主导地位,并定义了活性位点的整体构象;(iv)与Ser119形成强H键是有效抑制CYP3A4的先决条件; 和(v)系列中最强的抑制剂,R 1-苯基/ R 2-吲哚含8f(K s和IC 50即使在阻止基于CYP3A4的机制失活的条件下,其分别为0.08和0.43μM的浓度仍然比利托那韦的效力低。晶体学数据对于更好地理解和解释实验结果至关重要,并建议了如何进一步优化抑制剂的设计。


























 京公网安备 11010802027423号
京公网安备 11010802027423号