当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Understanding and Breaking Scaling Relations in Single-Site Catalysis: Methane to Methanol Conversion by FeIV═O
ACS Catalysis ( IF 12.9 ) Pub Date : 2018-01-05 00:00:00 , DOI: 10.1021/acscatal.7b03597 Terry Z. H. Gani 1 , Heather J. Kulik 1
ACS Catalysis ( IF 12.9 ) Pub Date : 2018-01-05 00:00:00 , DOI: 10.1021/acscatal.7b03597 Terry Z. H. Gani 1 , Heather J. Kulik 1
Affiliation
|
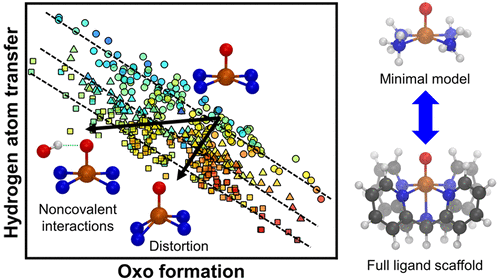
Computational high-throughput screening is an essential tool for catalyst design, limited primarily by the efficiency with which accurate predictions can be made. In bulk heterogeneous catalysis, linear free energy relationships (LFERs) have been extensively developed to relate elementary step activation energies, and thus overall catalytic activity, back to the adsorption energies of key intermediates, dramatically reducing the computational cost of screening. The applicability of these LFERs to single-site catalysts remains unclear, owing to the directional, covalent metal–ligand bonds and the broader chemical space of accessible ligand scaffolds. Through a computational screen of nearly 500 model Fe(II) complexes for CH4 hydroxylation, we observe that (1) tuning ligand field strength yields LFERs by comparably shifting energetics of the metal 3d levels that govern the stability of different intermediates and (2) distortion of the metal coordination geometry breaks these LFERs by increasing the splitting between the dxz/dyz and dz2 metal states that govern reactivity. Thus, in single-site catalysts, low Brønsted–Evans–Polanyi slopes for oxo formation, which would limit peak turnover frequency achievable through ligand field tuning alone, can be overcome through structural distortions achievable in experimentally characterized compounds. Observations from this screen also motivate the placement of strong HB donors in targeted positions as a scaffold-agnostic strategy for further activity improvement. More generally, our findings motivate broader variation of coordination geometries in reactivity studies with single-site catalysts.
中文翻译:

了解和断裂缩放关系在单点催化:甲烷甲醇转化率的Fe IV = O
计算高通量筛选是催化剂设计的重要工具,主要受到可进行准确预测的效率的限制。在本体非均相催化中,线性自由能关系(LFER)已得到广泛发展,以将基本步骤活化能以及总体催化活性与关键中间体的吸附能相关联,从而大大降低了筛选的计算成本。由于方向性,共价金属-配体键以及可利用的配体支架的化学空间较宽,这些LFER在单中心催化剂上的适用性仍不清楚。通过近500种CH 4模型Fe(II)配合物的计算屏幕羟基化作用,我们观察到(1)调节配体场强,通过比较控制不同中间体稳定性的金属3d能级移动能,产生LFERs;(2)金属配位几何形状的畸变通过增加d之间的分裂而打破了这些LFERs。xz / d yz和d z 2控制反应性的金属态。因此,在单中心催化剂中,低羰基形成的Brønsted–Evans–Polanyi低斜率会限制仅通过配体场调节可达到的峰转换频率,而可通过在实验表征的化合物中实现的结构变形来克服。从该屏幕上观察到的结果还激发了将强力HB供体放置在目标位置上的方法,以此作为与脚手架无关的策略,以进一步改善活动。更普遍地,我们的发现促使在单中心催化剂的反应性研究中配位几何形状的变化更加广泛。
更新日期:2018-01-05
中文翻译:
了解和断裂缩放关系在单点催化:甲烷甲醇转化率的Fe IV = O
计算高通量筛选是催化剂设计的重要工具,主要受到可进行准确预测的效率的限制。在本体非均相催化中,线性自由能关系(LFER)已得到广泛发展,以将基本步骤活化能以及总体催化活性与关键中间体的吸附能相关联,从而大大降低了筛选的计算成本。由于方向性,共价金属-配体键以及可利用的配体支架的化学空间较宽,这些LFER在单中心催化剂上的适用性仍不清楚。通过近500种CH 4模型Fe(II)配合物的计算屏幕羟基化作用,我们观察到(1)调节配体场强,通过比较控制不同中间体稳定性的金属3d能级移动能,产生LFERs;(2)金属配位几何形状的畸变通过增加d之间的分裂而打破了这些LFERs。xz / d yz和d z 2控制反应性的金属态。因此,在单中心催化剂中,低羰基形成的Brønsted–Evans–Polanyi低斜率会限制仅通过配体场调节可达到的峰转换频率,而可通过在实验表征的化合物中实现的结构变形来克服。从该屏幕上观察到的结果还激发了将强力HB供体放置在目标位置上的方法,以此作为与脚手架无关的策略,以进一步改善活动。更普遍地,我们的发现促使在单中心催化剂的反应性研究中配位几何形状的变化更加广泛。

























 京公网安备 11010802027423号
京公网安备 11010802027423号