当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Theoretical Models for Quantitative Description of the Acid–Base Equilibria of the 5,6-Substituted Uracils
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2017-12-28 00:00:00 , DOI: 10.1021/acs.jpca.7b09330 Margarita G. Ilyina 1, 2 , Edward M. Khamitov 1, 2, 3 , Sergey P. Ivanov 3 , Akhat G. Mustafin 1, 3 , Sergey L. Khursan 3
The Journal of Physical Chemistry A ( IF 2.9 ) Pub Date : 2017-12-28 00:00:00 , DOI: 10.1021/acs.jpca.7b09330 Margarita G. Ilyina 1, 2 , Edward M. Khamitov 1, 2, 3 , Sergey P. Ivanov 3 , Akhat G. Mustafin 1, 3 , Sergey L. Khursan 3
Affiliation
|
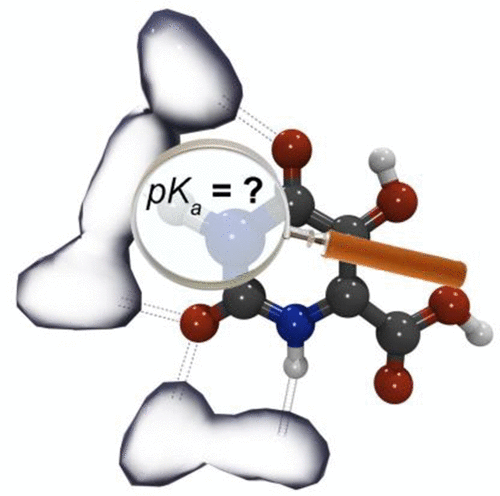
The acidities of 18 5,6-substituted uracils have been numerically estimated as pKa values in terms of three theoretical models. The first scheme includes the calculation of the gas-phase acidity of uracil with the G3MP2B3 method and taking into account the solvent effect using the polarizable continuum approximation PCM(SMD)-TPSS/aug-cc-pVTZ. The second model is one step and implies calculation of the free Gibbs energies of the hydrate complex of uracil (and its anion) with 5 water molecules by the TPSS/aug-cc-pVTZ method. This model accounts for the solvent effect corresponding to both specific and nonspecific solvation. The third scheme required high time and computational resources and includes the strong features of the two previous schemes. Here, the theoretical estimation of pKa is performed by the CBS-QB3 composite method. As in the second approach, both specific (as pentahydrate) and nonspecific solvent effects are determined. We have analyzed the advantages and model restrictions of the considered schemes for the pKa calculations. All models have systematic errors, which have been corrected with the linear empirical regression relations. In the presented model, the absolute mean deviations of the pKa values of uracils dissociating via the N1–H bonds diminish to 0.25, 0.28, and 0.23 pKa units (respectively, for I, II, and III models), which corresponds to ∼0.3 kcal/mol on the energy scale. The applicability of our computational schemes to uracils dissociating via N3–H, O–H (orotic acids) and C–H bonds is discussed.
中文翻译:

5,6-取代尿嘧啶的酸碱平衡定量描述的理论模型
根据三个理论模型,已通过数值将18个5,6-取代的尿嘧啶的酸度估计为p K a值。第一种方案包括使用G3MP2B3方法计算尿嘧啶的气相酸度,并使用可极化连续体近似PCM(SMD)-TPSS / aug-cc-pVTZ考虑溶剂效应。第二个模型是一个步骤,它意味着通过TPSS / aug-cc-pVTZ方法计算尿嘧啶(及其阴离子)与5个水分子的水合物的自由Gibbs能量。该模型考虑了对应于特异和非特异溶剂化的溶剂效应。第三种方案需要大量时间和计算资源,并且包括前两种方案的强大功能。这里,p K的理论估计a通过CBS-QB3复合方法执行。与第二种方法一样,确定了特定的(作为五水合物)和非特定的溶剂效果。我们已经分析了用于p K a计算的方案的优势和模型限制。所有模型都有系统误差,已通过线性经验回归关系进行了校正。在提出的模型中,通过N1-H键解离的尿嘧啶的p K a值的绝对平均偏差减小为0.25、0.28和0.23 p K a单位(分别针对I,II和III模型),在能量规模上相当于〜0.3 kcal / mol。讨论了我们的计算方案对通过N3–H,OH–(乳清酸)和C–H键解离的尿嘧啶的适用性。
更新日期:2017-12-28
中文翻译:
5,6-取代尿嘧啶的酸碱平衡定量描述的理论模型
根据三个理论模型,已通过数值将18个5,6-取代的尿嘧啶的酸度估计为p K a值。第一种方案包括使用G3MP2B3方法计算尿嘧啶的气相酸度,并使用可极化连续体近似PCM(SMD)-TPSS / aug-cc-pVTZ考虑溶剂效应。第二个模型是一个步骤,它意味着通过TPSS / aug-cc-pVTZ方法计算尿嘧啶(及其阴离子)与5个水分子的水合物的自由Gibbs能量。该模型考虑了对应于特异和非特异溶剂化的溶剂效应。第三种方案需要大量时间和计算资源,并且包括前两种方案的强大功能。这里,p K的理论估计a通过CBS-QB3复合方法执行。与第二种方法一样,确定了特定的(作为五水合物)和非特定的溶剂效果。我们已经分析了用于p K a计算的方案的优势和模型限制。所有模型都有系统误差,已通过线性经验回归关系进行了校正。在提出的模型中,通过N1-H键解离的尿嘧啶的p K a值的绝对平均偏差减小为0.25、0.28和0.23 p K a单位(分别针对I,II和III模型),在能量规模上相当于〜0.3 kcal / mol。讨论了我们的计算方案对通过N3–H,OH–(乳清酸)和C–H键解离的尿嘧啶的适用性。

























 京公网安备 11010802027423号
京公网安备 11010802027423号