当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Nonadiabatic Ab Initio Molecular Dynamics with the Floating Occupation Molecular Orbital-Complete Active Space Configuration Interaction Method
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2017-12-22 00:00:00 , DOI: 10.1021/acs.jctc.7b00958 Daniel Hollas 1 , Lukáš Šištík 1 , Edward G. Hohenstein 2, 3 , Todd J. Martínez 4, 5 , Petr Slavíček 1, 6
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2017-12-22 00:00:00 , DOI: 10.1021/acs.jctc.7b00958 Daniel Hollas 1 , Lukáš Šištík 1 , Edward G. Hohenstein 2, 3 , Todd J. Martínez 4, 5 , Petr Slavíček 1, 6
Affiliation
|
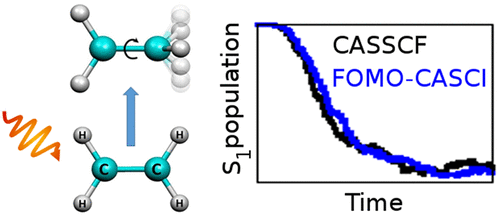
We show that the floating occupation molecular orbital complete active space configuration interaction (FOMO-CASCI) method is a promising alternative to the widely used complete active space self-consistent field (CASSCF) method in direct nonadiabatic dynamics simulations. We have simulated photodynamics of three archetypal molecules in photodynamics: ethylene, methaniminium cation, and malonaldehyde. We compared the time evolution of electronic populations and reaction mechanisms as revealed by the FOMO-CASCI and CASSCF approaches. Generally, the two approaches provide similar results. Some dynamical differences are observed, but these can be traced back to energetically minor differences in the potential energy surfaces. We suggest that the FOMO-CASCI method represents, due to its efficiency and stability, a promising approach for direct ab initio dynamics in the excited state.
中文翻译:

漂浮职业分子轨道-完全活性空间构型相互作用法的非绝热从头算分子动力学
我们表明,在直接非绝热动力学模拟中,浮动占领分子轨道完全活动空间构型相互作用(FOMO-CASCI)方法是广泛使用的完全活动空间自洽场(CASSCF)方法的有前途的替代方法。我们在光动力学中模拟了三个原型分子的光动力学:乙烯,甲烷亚胺阳离子和丙二醛。我们比较了FOMO-CASCI和CASSCF方法揭示的电子种群的时间演化和反应机理。通常,这两种方法提供相似的结果。观察到一些动态差异,但可以追溯到势能面在能量上的微小差异。我们建议,由于FOMO-CASCI方法的效率和稳定性,它代表了
更新日期:2017-12-22
中文翻译:
漂浮职业分子轨道-完全活性空间构型相互作用法的非绝热从头算分子动力学
我们表明,在直接非绝热动力学模拟中,浮动占领分子轨道完全活动空间构型相互作用(FOMO-CASCI)方法是广泛使用的完全活动空间自洽场(CASSCF)方法的有前途的替代方法。我们在光动力学中模拟了三个原型分子的光动力学:乙烯,甲烷亚胺阳离子和丙二醛。我们比较了FOMO-CASCI和CASSCF方法揭示的电子种群的时间演化和反应机理。通常,这两种方法提供相似的结果。观察到一些动态差异,但可以追溯到势能面在能量上的微小差异。我们建议,由于FOMO-CASCI方法的效率和稳定性,它代表了


























 京公网安备 11010802027423号
京公网安备 11010802027423号