当前位置:
X-MOL 学术
›
Chem. Bio. Drug Des.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Synthesis, adenosine receptor binding and molecular modelling studies of novel thieno[2,3‐d]pyrimidine derivatives
Chemical Biology & Drug Design ( IF 3 ) Pub Date : 2017-12-22 , DOI: 10.1111/cbdd.13155 Pran Kishore Deb 1, 2 , Raghuprasad Mailavaram 3 , Balakumar Chandrasekaran 1 , Venkata Rao Kaki 1 , Rajwinder Kaur 1 , Sonja Kachler 4 , Karl-Norbert Klotz 4 , Raghuram Rao Akkinepally 5
Chemical Biology & Drug Design ( IF 3 ) Pub Date : 2017-12-22 , DOI: 10.1111/cbdd.13155 Pran Kishore Deb 1, 2 , Raghuprasad Mailavaram 3 , Balakumar Chandrasekaran 1 , Venkata Rao Kaki 1 , Rajwinder Kaur 1 , Sonja Kachler 4 , Karl-Norbert Klotz 4 , Raghuram Rao Akkinepally 5
Affiliation
|
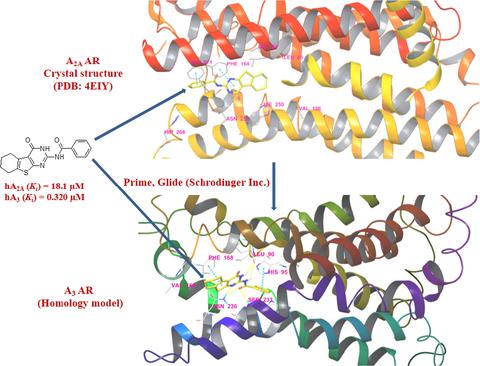
A series of new molecules containing a thieno[2,3‐d]pyrimidine scaffold was synthesized and characterized by adopting an efficient synthetic scheme. The effect of a free or substituted amino group at 2‐position as well as an oxo‐group, imidazole or 1,2,4‐triazole ring at 4‐position of the scaffold on the affinity and selectivity towards adenosine receptors (ARs) was evaluated. Compounds 17–19 with a free amino group at 2‐position along with the presence of an imidazole/1,2,4‐triazole ring at 4‐position of the scaffold showed selective binding affinities for hA2A AR, whereas carbamoylation of the amino group at 2‐position (in the presence of an oxo‐group at 4‐position of the scaffold) increased the affinity and selectivity of certain compounds (7–10) for hA3 AR. Molecular dynamic simulation study of one of the most active compound 8 (Ki hA1 > 30 μm, hA2A = 0.65 μm, and hA3 = 0.124 μm) revealed the role of important amino acid residues for imparting good affinity towards hA3 and hA2A ARs. Molecular docking studies were carried out for other compounds using the crystal structure of hA2A AR and a homology model of hA3 AR to rationalize their structure–activity relationships. The molecular docking results were in agreement with the experimental binding affinity data of ARs.
中文翻译:

新型噻吩并[2,3-d]嘧啶衍生物的合成,腺苷受体结合和分子建模研究
合成了一系列新的含有噻吩并[2,3- d ]嘧啶骨架的分子,并采用有效的合成方案对其进行了表征。在支架的2位上的游离或取代的氨基以及在4位上的氧代基,咪唑或1,2,4-三唑环对腺苷受体(ARs)的亲和力和选择性的影响是评估。化合物17-19在2位带有一个游离氨基,并且在支架的4位上存在一个咪唑/ 1,2,4-三唑环,对hA 2A AR表现出选择性结合亲和力,而氨基甲氨甲酰化2位的基团(在支架4位的基团存在一个氧代基团的情况下)提高了某些化合物的亲和力和选择性(7–10)适用于hA 3 AR。最活跃的化合物之一的分子动力学模拟研究8(ķ我HA 1 > 30μ米,HA 2A = 0.65μ米,和HA 3 = 0.124μ米)揭示的重要氨基酸残基的作用,为赋予朝良好的亲和性hA 3和hA 2A AR。使用hA 2A AR的晶体结构和hA 3的同源性模型对其他化合物进行了分子对接研究通过AR合理化他们的结构-活动关系。分子对接结果与AR的实验结合亲和力数据一致。
更新日期:2017-12-22
中文翻译:
新型噻吩并[2,3-d]嘧啶衍生物的合成,腺苷受体结合和分子建模研究
合成了一系列新的含有噻吩并[2,3- d ]嘧啶骨架的分子,并采用有效的合成方案对其进行了表征。在支架的2位上的游离或取代的氨基以及在4位上的氧代基,咪唑或1,2,4-三唑环对腺苷受体(ARs)的亲和力和选择性的影响是评估。化合物17-19在2位带有一个游离氨基,并且在支架的4位上存在一个咪唑/ 1,2,4-三唑环,对hA 2A AR表现出选择性结合亲和力,而氨基甲氨甲酰化2位的基团(在支架4位的基团存在一个氧代基团的情况下)提高了某些化合物的亲和力和选择性(7–10)适用于hA 3 AR。最活跃的化合物之一的分子动力学模拟研究8(ķ我HA 1 > 30μ米,HA 2A = 0.65μ米,和HA 3 = 0.124μ米)揭示的重要氨基酸残基的作用,为赋予朝良好的亲和性hA 3和hA 2A AR。使用hA 2A AR的晶体结构和hA 3的同源性模型对其他化合物进行了分子对接研究通过AR合理化他们的结构-活动关系。分子对接结果与AR的实验结合亲和力数据一致。


























 京公网安备 11010802027423号
京公网安备 11010802027423号