当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Elucidating electrolyte decomposition under electron-rich environments at the lithium-metal anode
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2017-11-07 00:00:00 , DOI: 10.1039/c7cp06485c Luis E. Camacho-Forero 1, 2, 3, 4 , Perla B. Balbuena 1, 2, 3, 4, 5
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2017-11-07 00:00:00 , DOI: 10.1039/c7cp06485c Luis E. Camacho-Forero 1, 2, 3, 4 , Perla B. Balbuena 1, 2, 3, 4, 5
Affiliation
|
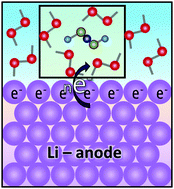
The lithium metal anode is one of the key components of the lithium–sulfur (Li–S) batteries, which are considered one of the most promising candidates for the next generation of battery systems. However, one of the main challenges that have prevented Li-metal anodes from becoming feasible to be used in commercial batteries is the continuous decomposition of the electrolyte due to its high reactivity, which leads to the formation of solid–electrolyte interphase (SEI) layers. The properties of the SEI can dramatically affect the performance of the batteries. Thus, a rigorous understanding of the electrolyte decomposition is crucial to elucidate improvements in performance of the Li–S technology. In this work, using density functional theory (DFT) and ab initio molecular dynamics simulations (AIMD), we investigate the effect of electron-rich environments on the decomposition mechanism of electrolyte species in pure 1,2-dimethoxyethane (DME) solvent and 1 M lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) and lithium bis(fluorosulfonyl)imide (LiFSI) salt solutions. It is found that systems with pure DME require an average environment of at least ∼0.9 |e| per molecule for a DME to decompose into CH3O− and C2H42−via a 4-electron transfer. In the case of mixtures, the salts are very prone to react with any excess of electrons. In addition, DME dehydrogenation due to reactions with fragments coming from the salt decompositions was detected. Formation of oligomer anionic species from DME and salt fragments were also identified from the AIMD simulations. Finally, the thermodynamics and kinetics of the most relevant electrolyte decomposition reactions were characterized. DME decomposition reactions predicted from the AIMD simulations were found to be thermodynamically favorable under exposure to Li atoms and/or by reactions with salt fragments. In most cases, these reactions were shown to have low to moderate activation barriers.
中文翻译:

阐明锂金属阳极在富电子环境下的电解质分解
锂金属阳极是锂硫(Li–S)电池的关键组件之一,被认为是下一代电池系统最有希望的候选者之一。但是,阻止锂金属阳极成为可行的商业电池的主要挑战之一是由于电解质的高反应性导致电解质的连续分解,这导致形成了固体-电解质中间相(SEI)层。SEI的特性会极大地影响电池的性能。因此,对电解质分解的严格了解对于阐明Li-S技术性能的提高至关重要。在这项工作中,使用密度泛函理论(DFT)和从头算分子动力学模拟(AIMD),我们研究了富电子环境对纯1,2-二甲氧基乙烷(DME)溶剂和1 M双(三氟甲磺酰基)酰亚胺锂(LiTFSI)和双(氟磺酰基)酰亚胺(LiFSI)盐溶液。发现具有纯DME的系统需要的平均环境至少约为0.9 | |。e | 每分子用于DME分解成CH 3 ö -和C 2 H ^ 4 2-经由4电子转移。在混合物的情况下,盐非常容易与任何过量的电子反应。另外,检测到由于与盐分解产生的碎片反应而引起的DME脱氢。还从AIMD模拟中确定了由DME和盐片段形成的低聚物阴离子物种。最后,对最相关的电解质分解反应的热力学和动力学进行了表征。发现由AIMD模拟预测的DME分解反应在暴露于Li原子下和/或与盐片段反应在热力学上是有利的。在大多数情况下,这些反应显示有低至中度的激活屏障。
更新日期:2017-11-22
中文翻译:
阐明锂金属阳极在富电子环境下的电解质分解
锂金属阳极是锂硫(Li–S)电池的关键组件之一,被认为是下一代电池系统最有希望的候选者之一。但是,阻止锂金属阳极成为可行的商业电池的主要挑战之一是由于电解质的高反应性导致电解质的连续分解,这导致形成了固体-电解质中间相(SEI)层。SEI的特性会极大地影响电池的性能。因此,对电解质分解的严格了解对于阐明Li-S技术性能的提高至关重要。在这项工作中,使用密度泛函理论(DFT)和从头算分子动力学模拟(AIMD),我们研究了富电子环境对纯1,2-二甲氧基乙烷(DME)溶剂和1 M双(三氟甲磺酰基)酰亚胺锂(LiTFSI)和双(氟磺酰基)酰亚胺(LiFSI)盐溶液。发现具有纯DME的系统需要的平均环境至少约为0.9 | |。e | 每分子用于DME分解成CH 3 ö -和C 2 H ^ 4 2-经由4电子转移。在混合物的情况下,盐非常容易与任何过量的电子反应。另外,检测到由于与盐分解产生的碎片反应而引起的DME脱氢。还从AIMD模拟中确定了由DME和盐片段形成的低聚物阴离子物种。最后,对最相关的电解质分解反应的热力学和动力学进行了表征。发现由AIMD模拟预测的DME分解反应在暴露于Li原子下和/或与盐片段反应在热力学上是有利的。在大多数情况下,这些反应显示有低至中度的激活屏障。

























 京公网安备 11010802027423号
京公网安备 11010802027423号